Comparação de sequências
|
|
|
- Madalena Bergler Peres
- 6 Há anos
- Visualizações:
Transcrição

1 Comparação de sequências João Carlos Setubal IQ-USP /22/2016 J. C. Setubal 1
2 Motivação

3 Ciccarelli et al, Science, 2006
4 Como foi possível criar tal árvore? Determinar genes que são compartilhados por todos os seres vivos O que é compartilhar? Gene x 1 em organismo A tem função α Gene x 2 em organismo B é homólogo e tem a mesma função α Então x 1 e x 2 são o mesmo gene x e portanto ele é compartilhado por A e B Quais genes são esses?

5
6 Questões Onde podemos achar as sequências dos genes? Como determinar compartilhamento? Como preparar esses dados para construir uma árvore? Como construir uma árvore? Como saber se ela esta correta? Algo novo desde 2006?

7 Fig. 1. Overview of the procedure. F D Ciccarelli et al. Science 2006;311: Published by AAAS
8 Respostas Onde podemos achar as sequências dos genes? Bancos de dados públicos (NCBI) BLAST Como determinar compartilhamento? Através de comparação de sequências Como preparar esses dados para construir uma árvore? Alinhamento múltiplo concatenado Como construir uma árvore? Métodos de reconstrução filogenética Como saber se ela esta correta? Inferência é um termo melhor do que construção Argumentos probabilísticos Transferência Horizontal (Lateral) de Genes Algo novo desde 2006? Next Generation Sequencing (NGS) Comparação de genomas completos
9 Por que comparar sequências? Achar similaridades Dadas 2 sequências, quão parecidas elas são? DNA e proteína Buscas em banco de dados Achar quais sequências do banco são parecidas com minha sequência-consulta Consulta (query) é tipicamente uma sequência nova google
10 Motivação (cont.) Construir famílias de proteínas Saber quais organismos tem membros da família Determinar uma assinatura para a família Construir filogenias Entender a história evolutiva de genes e organismos
11 Premissas Em geral buscamos sequências aparentadas Sequências aparentadas são similares aparentadas = homólogas Descendem de um mesmo ancestral Descendentes sofreram mutações ao longo do tempo
12 Alinhamento de DNA GTGGTGGCCTACGAAGGT GTAGTGCCTTCGAAGGGT
13 Como avaliar um alinhamento? Sistema de pontuação Match: +1 Mismatch: 1
14 Pontuação do alinhamento GTGGTGGCCTACGAAGGT GTAGTGCCTTCGAAGGGT = 4
15 É possível melhorar o alinhamento? Sim Pela introdução de espaços
16 Alinhamento com espaços GTGGTGGCCTACGAA-GGT GTAGTG-CCTTCGAAGGGT
17 Sistema de pontuação com espaços Match: +1 Mismatch: 1 Espaço: 2 (Buraco: sequência de espaços) Em inglês: gaps
18 Pontuação do alinhamento GTGGTGGCCTACGAA-GGT GTAGTG-CCTTCGAAGGGT = 9
19 Justificativa para o sistema de pontuação Matches tem que ser recompensados (> 0) Mismatches e espaços tem que ser penalizados (< 0) Mismatches representam substituições Mutações (ocorrem com frequência) Podem não trazer letalidade Espaços representam inserções ou remoções Mais prováveis de causarem letalidade Alteram quadro de leitura Ocorrem com muito menos frequência
20 Alinhamentos ótimos São os alinhamentos de pontuação máxima similaridade entre duas sequências É o valor da pontuação do alinhamento ótimo No exemplo anterior Similaridade = 9
21 Comparação de sequências de aminoácidos
22 Pontuação de alinhamento de proteínas % de identidade é uma medida simples mas válida de similaridade de sequências de proteínas
23 Aminoácidos se dividem em famílias Hidrofóbicos Ala, Val, Phe, Pro, Met, Ile, Leu Com carga Asp, Glu, Lys, Arg Polares Gly Ser, Thr, Tyr, His, Cys, Asn, Gln Trp

24
25 Mutações e proteínas Substituições que não alteram a estrutura da proteína tendem a ser preservadas durante a evolução A troca de um aminoácido de uma família por outro da mesma família em geral cai nessa categoria (Indels podem ter consequências mais drásticas) Então: como avaliar mismatches?
26 Matriz de substituição de amino ácidos BLOSUM62 Fonte: NCBI
27 Pontuação leva em conta a matriz Match: blosum62(i,i) sempre positivo Mismatch: blosum62(i,j) positivo, nulo, negativo Espaço: 2
28 Exercício Se fosse necessário criar uma matriz de substituição para nucleotídeos, que critério poderia ser usado? Critério = propriedades dos diferentes nucleotídeos Purinas (A, G) e pirimidinas (C, T) na prática isto não é feito
29 Como obter alinhamentos ótimos? Precisamos de um algoritmo Algoritmos são diferentes de programas Algoritmo é um método abstrato Programa é uma encarnação física (numa linguagem de programação) de um algoritmo Um programa pode ser executado num computador Dizemos que um certo algoritmo foi implementado em tal ou qual linguagem Quem escrever algoritimo na prova perde 3 pontos!
30 Algoritmo para achar alinhamentos ótimos de DNA Desenvolvido com a técnica de Programação dinâmica Técnica desenvolvida na década de 1950 por Richard Bellman Não é programação e não é dinâmica!
31 Programação Dinâmica PD se usa para problemas que tem uma estrutura de subproblemas Num alinhamento com sequências s e t um subproblema é qualquer alinhamento entre s e t tal que s = um prefixo de s e t = um prefixo de t Um prefixo 9/22/2016 J. C. Setubal 31
32 Ideia básica da PD Achar soluções de subproblemas e armazená-las numa tabela (matriz) Para achar a solução ótima: Ir achando as soluções na direção dos subproblemas menores para os maiores Último elemento da tabela a ser preenchido contém a solução do problema completo
33 j i t G A T C 0 s 1 G 2 T 3 C
34 Preenchimento da tabela Este processo precisa começar com o menor subproblema possível. Qual seria? Quando pelo menos uma das sequências é vazia Inicialização: Alinhar s com cadeia vazia e alinhar t com cadeia vazia
35 j i t G A T C 0 s G -2 2 T -4 3 C -6
36 Continuação Alinhar caracter X com caracter Y 3 possibilidades X com Y Aplicar pontuação respectiva, dependendo se for DNA ou proteína X com espaço Cobrar -2 Y com espaço Cobrar -2
37 Preenchimento de (1,1) Significa determinar qual é o melhor alinhamento dos prefixos de s e t com apenas um caracter cada um Alternativas G- -G G -G G- G Todos eles usam valores determinados na inicialização
38 j i t G A T C 0 s G T -4 3 C -6
39 j i t G A T C 0 s G T C
40 Exercícios Inventar duas sequências de DNA curtas e rodar (na mão) o algoritmo de PD Para se auto-corrigir: Sequence-Alignment-using-Dynamic-Programming-A (busque dna sequence alignment code project) Demo parece segura Exige registro Tem código fonte
41 Complexidade computacional de PD Queremos saber quanto tempo gasta o algoritmo Mas algoritmos são abstratos, não estão associados a computadores físicos; o que significa tempo? Então contamos número de operações elementares Operações aritméticas Uso de posições da memória Etc Porém queremos apenas algo aproximado Uma ordem de grandeza
42 Complexidade computacional Queremos expressar o número de operações como uma função matemática do tamanho das entradas Por exemplo n (linear) n 2 (quadrático) n log n Vamos desprezar constantes (n e 30n dão na mesma) Só nos interessa o termo de maior grau (desprezar os demais) 7n n log n vira apenas n 2 Vamos usar a notação O(f(n)) para denotar tudo isso
43 Complexidade computacional de PD A matriz tem tamanho n+1 por m+1 Todos os elementos da matriz precisam ser preenchidos Supondo tempo constante para o preenchimento n+1 m+1 = nm + n + m + 1 O(nm) Se n m, O(n 2 ) Quadrático Memória: quadrático também
44
45 Penalização de espaços pode ser mais sofisticada GTGGTGGCCTACGAAGGT GTGGTCGC---CGAAGGT GTGGTGGCC-ACGAAGGT GT-GTCGCCTACGA-GGT No sistema de pontuação apresentado, k espaços consecutivos (um buraco ou gap) custam o mesmo que k espaços separados Seria melhor distinguir os dois casos
46 Penalização de espaços feita por uma função matemática k = número de espaços p(k) = a + bk p(k) é subtraído do score a = custo para abrir um buraco b = custo para continuar um buraco Por exemplo: p(k) = 2 + k 5 espaços consecutivos custam 7 5 espaços separados custam 15 Compare com a função implícita do sistema simples: p(k) = 2k
47 Algoritmo de PD com penalização de buracos por função afim O algoritmo é mais complexo São necessárias 3 tabelas ao invés de 1 Mas a complexidade permanece a mesma (quadrática) Algoritmo de Smith-Waterman
48 Smith-Waterman
49 Se a função for genérica [ex. p(k) = a + b log k)], então a complexidade passa para O(n 3 ) Algoritmo de Needleman-Wunsch
50 Queremos descobrir sequências aparentadas Aparentadas = ancestral comum = homólogas Alinhamentos biologicamente relevantes Nota máxima por si só não nos informa sobre parentesco Alinhamentos de nota máxima não necessariamente correspondem a alinhamentos biologicamente relevantes Como fazer?
51 Bancos de sequências Situação típica Tenho uma sequência consulta Quero saber se existem sequências já publicadas que são parentes dela Tenho que fazer uma busca em bancos de sequências
52 Bancos de sequências Resultado do sequenciamento em geral é publicado bancos de dados de sequências Na verdade catálogos Mais importante: GenBank Mantido pelo National Center for Biotechnological Information NCBI

53

54 UniProt

55 Estatística de alinhamentos Com um banco, temos uma população de sequências Com essa população, posso criar uma teoria estatística que vai me permitir separar os alinhamentos estatisticamente significativos daqueles obtidos por mero acaso Diremos que os alinhamentos estatisticamente significativos são biologicamente relevantes A significância estatística precisa ser quantificada: e-value
56 E-value Teoria de Karlin e Altschul Calcula o e-value (expect value) de um alinhamento E = Kmne λs m e n são os tamanhos das sequências S é a pontuação K e λ são parâmetros Um banco de sequências pode ser tratado como uma longa sequência de tamanho n A fórmula dá o número de alinhamentos que se esperaria obter com pontuação pelo menos S ao acaso
57 e-value Não é uma probabilidade Pode resultar maior do que 1 Mas em geral os alinhamentos biologicamente relevantes tem e-value < 10 5 Para valores assim ou menores, o e-value se comporta como uma probabilidade p-values e e-values P = 1 e E
58 E-value depende do tamanho do banco Não se pode comparar diretamente e-values obtidos de consultas a bancos diferentes Mas existe uma fórmula de conversão Dado o e-value contra banco X, é possível saber qual seria o e-value contra banco Y Essa mesma fórmula pode ser usada para dar o e- value para comparação de apenas duas sequências entre si (supondo que Y seja genbank)

59
60 Programação dinâmica é cara Especialmente quando Comparação contra muitas sequências Buscas em banco de dados Comparação de muitas sequências entre si Todas contra todas Alternativa: BLAST Basic Local Alignment Search Tool
61 BLAST Altschul et al., 1990, 1997 Cerca de 75 mil citações (10/2014) Programa mais citado em ciência (mas não são os papers mais citados; o mais citado tem 305 mil citações) Heurística Não tem garantia de que sempre consegue achar os alinhamentos de pontuação máxima Sacrifica garantia de otimalidade por velocidade Mas na vasta maioria das vezes tais alinhamentos são de fato encontrados Reporta e-values (É possível fazer cálculo de e-values com PD)

62 BLAST Acha alinhamentos locais global
63 É útil pensar na matriz de PD como se fosse um dotplot j i t G A T C 0 s G T C
64 Um alinhamento local é um trecho de células consecutivas num dotplot
65 Pode haver vários alinhamentos locais
66 Alinhamento global
67 Exercício Como modificar o algoritmo de PD para que ele encontre o melhor alinhamento local? Dica: comece definindo as características do melhor alinhamento local
68 Alinhamento local com PD Um alinhamento global pode ter nota negativa um alinhamento local nunca pode ter nota negativa Pois o alinhamento entre sequências vazias tem nota zero bons alinhamentos locais são trechos com notas positivas na matriz de PD
69 Implementação 1. Inicialização da coluna zero e da linha zero com zeros 2. Ao preencher um elemento da matriz, fazer a operação de máximo, mas nunca deixar que o valor escolhido seja negativo Valor max (M[i-1,j], M[i, j-1], M[i-1,j-1], 0) 3. Ao final: procurar o elemento da matriz de valor máximo 4. Para recuperar o alinhamento: percorrer de trás para diante até chegar em zero
70 De onde vem a eficiência de BLAST? BLAST busca trechos parecidos (palavras ou words) entre as sequências = alinhamentossemente Para nt, esses alinhamentos tem que ser exatos Para aa, esses alinhamentos tem que ter nota positiva Estende esses alinhamentos-semente
71 Crédito: I. Lobo
72 Exemplo de alinhamento ótimo que BLAST não encontraria Suponha nt e tamanho mínimo de palavras = 4 GTG-TGGCCTA-GAAGGT GTGGTGG-CTACGAA-GT
73 Características de BLAST Tamanho default das palavras DNA: 11 nt Proteínas: 3 aa Reporta bit score, raw score, e-value, identidades, positivos, buracos

74

75

76 Buscando no GenBank

77 Lista de hits
78 Sabores de BLAST Subject nucleotídeos aminoácidos Query nucleotídeos BLASTN TBLASTX BLASTX aminoácidos TBLASTN BLASTP Também: megablast, psi-blast, phi-blast, delta-blast JC Setubal 78
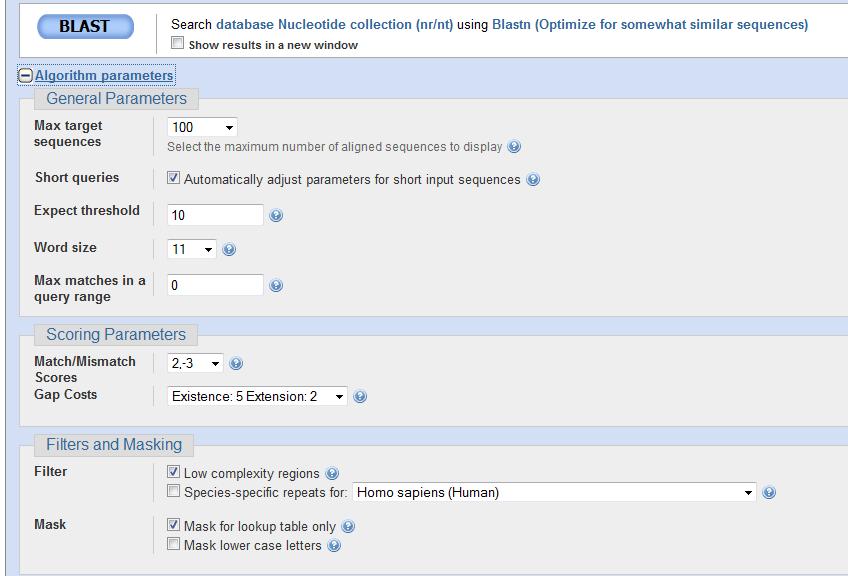
79 Parâmetros de BLASTn
80 Regiões de baixa complexidade Sequências com elementos repetitivos e que aparecem com frequência Exemplo em DNA AAAAAAA Exemplo em proteína AGNLLGRNVVVVGAG Uso do filtro é default Pode excluir alinhamentos relevantes

81 Parâmetros de BLASTp
82 Posso acreditar nos resultados do BLAST? 10-5 Falsos negativos Falsos positivos Não significante significante 1) Nem todos os alinhamentos estatisticamente significativos são biologicamente relevantes 2) Nem todos os alinhamentos que não são estatisticamente significantes não são relevantes

83 Exemplo do caso (1) Duas proteínas podem compartilhar um domínio e não serem relacionadas falsos positivos de BLAST Acontece quando as proteínas tem múltiplos domínios Pyruvate kinase

84 Referências Ian Korf, Mark Yandell, Joseph Bedell Artigo por Ingrid Lobo (Write Science Right) 2008 Nature Education

85
86 Novos programas Usearch [Edgar 2010] Até 400x mais rápido do que BLAST Com algum sacrifício de precisão Pauda [Huson e Xie, 2014] Blastx dos pobres x mais rápido do que blastx! Com mais sacrifício de precisão Diamond [Buchfink, Xie, Huson 2014] x mais rápido do que blastx! Sem sacrifício de precisão!
87 Alinhamento múltiplo

88 Alinhamento múltiplo Para construir filogenias é necessário criar AMs JC Setubal 88
89 Avaliação de AMs por soma de pares Numa coluna, podemos separar todos os pares de aminoácidos 1 com 2, 1 com 3, 1 com 4, etc 2 com 3, 2 com 4, etc A cada par corresponde uma nota na matriz BLOSUM62 A soma de todas as notas dos pares dá a nota da coluna A soma das notas das colunas dá a nota do alinhamento
90 Coluna de um alinhamento L I V V I BLOSUM62 L/L: 4 L/I: 2 L/V: 1 I/I: 4 I/V: 3 V/V: 4 L I V V I L I V V 3 3 I 26 Nota da coluna
 indicates positions which have a single, fully conserved residue A : (colon) indicates conservation between groups of strongly similar properties - scoring > 0.")
91 What do the consensus symbols mean in the alignment? An * (asterisk) indicates positions which have a single, fully conserved residue A : (colon) indicates conservation between groups of strongly similar properties - scoring > 0.5 in the Gonnet PAM 250 matrix A. (period) indicates conservation between groups of weakly similar properties - scoring =< 0.5 in the Gonnet PAM 250 matrix
92 Não existe padrão universalmente aceito para avaliar AMs Ou seja, não existe o equivalente de e-values em BLAST Diferentes programas produzem diferentes notas

93 Multiple Sequence Alignment 9/22/2016 J. C. Setubal 93
94 Sequências de entrada Dois conceitos importantes Homologia Família 9/22/2016 J. C. Setubal 94
95 Homologia Dois genes que tem um mesmo ancestral são homológos Freq. usado erroneamente com o sentido de similar Similaridade não implica necessariamente em homologia Asas: morcêgo e insetos (convergência) Às vezes a similaridade é (ou parece) baixa mas mesmo assim existe homologia Barbatana de baleia e braços em humanos Dois tipos de homologia Ortologia e paralogia
96 Ortólogos especiação 22 September 2016 JC Setubal 96
97 parálogos Figure by C. Lasher 22 September 2016 JC Setubal 97
98 In-parálogos Figure by C. Lasher 22 September 2016 JC Setubal 98
99 Homologia e função Seria bom se proteína homólogas tivessem mesma função Geralmente é o caso; mas nem sempre Parálogos estão mais sujeitos a desenvolver novas funções Neo-funcionalização Na prática Membros de uma mesma família de proteínas são homólogos e em geral tem mesma função Superfamílias e subfamílias
100 Família de proteínas Definição operacional Duas proteínas estão na mesma família se seus genes são homólogos ou (mais exigente) Duas proteínas estão na mesma família se seus genes são ortólogos Falar em proteínas homólogas é um certo abuso de linguagem: são os genes que são homólogos

101 Phylogenetic tree of the WHAMM proteins Kollmar et al. BMC Research Notes :88 doi: /
102 Colunas num AM devem ser homólogas O gene ancestral comum das sequências no AM também tinha aquela posição
103 Alinhar DNA ou aminoácidos? DNA: mais difícil garantir homologia nas colunas DNA é mais sensível, mas a 3a base de codons não é informativa Comparação com aminoácidos permite que proteínas mais distantes possam ser incluídas Há casos em que não dá para alinhar DNA (muita divergência) DNA é indicado quando as proteínas são todas idênticas ou quase idênticas Ex: cepas de uma bactéria 9/22/2016 J. C. Setubal 103
104 Algoritmo para alinhamento múltiplo de sequências Programação dinâmica Generalização de alinhamento 2-a-2 9/22/2016 J. C. Setubal 104

 O(n 3 ) Ω(2 k n k")
105 Generalização de PD para AM 2 sequências 3 sequências x y O(n 2 ) O(n 3 ) Ω(2 k n k )
106 Consequência Se PD para alinhamentos 2-a-2 já é caro para AM é ainda mais caro! Portanto todos os programas práticos para AM são heurísticas Não tem garantia de otimalidade (produzem aproximações)
107 Mesmo sendo heurísticas esses programas tem limitações As sequências de entrada: Não muito longas (menos do que ~10 kb) Não muitas (menos do que ~500) esses números variam dependendo do programa e do computador 9/22/2016 J. C. Setubal 107
108 Alinhamento progressivo Ideia: combinar alinhamentos de pares, iniciando com o par mais similar entre si Ir juntando os pares Dois estágios 1. constrói-se uma árvore-guia que determina a hierarquia de similaridade entre os pares 2. as sequências são adicionadas ao alinhamento num processo guiado pela árvore Seria melhor que AM e árvore fossem feitos simultaneamente Muito mais complicado de fazer com rigor
109 Programas para AM Muscle Edgar, R.C. (2004) Nucleic Acids Res. 32(5): MAFFT Katoh, Misawa, Kuma, Miyata 2002 (Nucleic Acids Res. 30: ) mafft.cbrc.jp/alignment/software/ ClustalW/X (antigos) Clustal Omega (novo) Sievers et al. Molecular Systems Biology (2011) 7: Outros: Probcons, Cobalt (NCBI), T-coffee 9/22/2016 J. C. Setubal 109

110
 Reference alignment of representative sequences of the p53/p63/p73 family, with the domain organization shown above (B) the alignment (AD: activation domain, Oligo: oligomerization, SAM: sterile")
111 Figure 1: An example benchmark alignment. (A) Reference alignment of representative sequences of the p53/p63/p73 family, with the domain organization shown above (B) the alignment (AD: activation domain, Oligo: oligomerization, SAM: sterile alpha motif). Colored blocks indicate conserved (C) regions. The grey regions correspond to sequence segments that could not be reliably aligned and white regions indicate (D) gaps in the alignment. (B) Different MSA programs produce different alignments, especially in the N-terminal region (boxe (E) d in red in A) containing rare motifs and a disordered proline-rich domain. Esquema comparativo de notas

112 Edição de alinhamentos Credit: R. Dixon 9/22/2016 J. C. Setubal 112
113 Edição de alinhamentos Algumas colunas podem não ser informativas No olho às vezes é possível ver alinhamentos locais melhores Edição manual Edição automática 9/22/2016 J. C. Setubal 113
114 Edição manual de AMs Jalview Waterhouse et al. Bioinformatics (9) Seaview Gouy M., Guindon S. & Gascuel O. (2010) Molecular Biology and Evolution 27(2):

115 JALVIEW 9/22/2016 J. C. Setubal 115

116
117 Edição automática de AMs GBLOCKS Castresana, J. (2000) Molecular Biology and Evolution 17, GUIDANCE Penn, O., Privman, E., Ashkenazy, H., Landan, G., Graur, D. and Pupko, T. (2010). GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Research, 2010 Jul 1; 38 (Web Server issue):w23-w28; doi: /nar/gkq443
118 Formatos de saída clustal, FASTA, MSF, NEXUS, PHYLIP
119 Alinhamento entre sequências longas Cromossomos inteiros O cromossomo típico de uma bactéria tem 4 Mbp Cromossomo de humanos: 300 Mbp Cromossomos e plasmídeos: replicons
120 22 September 2016 JC Setubal 120
121 BLAST não serve Computadores mesmo com dezenas de GB de RAM não dão conta de rodar BLAST para essas entradas Problema não é tempo; é memória RAM Outras abordagens são necessárias
122 O programa MUMmer Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res Jun 1;30(11): Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R September 2016 JC Setubal 122
123 Como MUMmer funciona It finds Maximal Unique Matches These are exact matches above a user-specified threshold that are unique Exact matches found are clustered and extended (using dynamic programming) Result is approximate matches Data structure for exact match finding: suffix tree Difficult to build but very fast Nucmer and promer Both very fast O(n + #MUMs), n = genome lengths 22 September 2016 JC Setubal 123
124 Árvore de sufixos para GTATCTAGG
125 Alinhamentos de replicons inteiros revelam rearranjos
126 Alinhamentos de pares de replicons completos Se as sequências fossem idênticas veríamos: B A 22 September 2016 JC Setubal 126
127 uma inversão A B C D A D C B 22 September 2016 JC Setubal 127
128 D B C A A B C D Such inversions seem to happen around 22 September 2016 the origin or terminus JC Setubal of replication 128
129 129
130 22 September 2016 JC Setubal 130
131
132 E. coli K12 Promer alignment Red: direct; green: reverse Xanthomonas axonopodis pv citri Both are proteobacteria! 22 September 2016 JC Setubal 132
133 Eisen JA, Heidelberg JF, White O, Salzberg SL. Evidence for JC symmetric Setubal chromosomal inversions around the replication origin in bacteria. Genome Biol. 2000;1(6):RESEARCH September
134 Alinhamento múltiplo de O programa MAUVE sequências longas Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res Jul;14(7): September 2016 JC Setubal 134
135 How MAUVE works Seed-and-extend hashing Seeds/anchors: Maximal Multiple Unique Matches of minimum length k Result: Local collinear blocks (LCBs) O(G 2 n + Gn log Gn), G = # genomes, n = average genome length 22 September 2016 JC Setubal 135
136 Alignment algorithm 1. Find Multi-MUMs 2. Use the multi-mums to calculate a phylogenetic guide tree 3. Find LCBs (subset of multi-mums; filter out spurious matches; requires minimum weight) 4. Recursive anchoring to identify additional anchors (extension of LCBs) 5. Progressive alignment (CLUSTALW) using guide tree 22 September 2016 JC Setubal 136

137 Main chromosome alignment MAUVE 22 September 2016 JC Setubal 137

138 Chromosome 2 alignment MAUVE 22 September 2016 JC Setubal 138

139 Chromosome alignment MAUVE Dugway RSA 493 RSA September 2016 JC Setubal 139

140 Genome Alignments MAUVE 22 September 2016 JC Setubal 140
141 Sumário de comparação de sequências Sequências curtas Sequências longas 2-a-2 Prog. Dinâmica Mummer 2-a-2 muitas vezes BLAST, usearch, diamond Mummer múltiplo Muscle, MAFFT Mauve, MUGSY
142 Comparação de conjuntos de genes Given a set of genomes, represented by their proteomes or sets of protein sequences Given homologous relationships (as given for example by orthomcl) Which genes are shared by genomes X and Y? Which genes are unique to genome Z? Venn or extended Venn diagrams 22 September 2016 JC Setubal 142
143 3-way genome comparison A B C 22 September 2016 JC Setubal 143

144

145 Diagrama de Venn para n = 6 Número de comparações é quadrático em n Número de regiões num diagrama de Venn = 2 n

146 Wulff et al. MPMI Vol. 27, No. 2, 2014, pp
147 Cômputo de famílias de proteínas 1. Verificar as similaridades entre as sequências a) Usando (por exemplo) BLAST + critérios 2. Representar as similaridades num grafo 3. Aplicar um algoritmo de clusterização sobre o grafo
148 Clusterização é necessária porque o grafo pode ser complexo a b c
149 Resultado da clusterização Matriz com genes nas colunas (g j ), genomas nas linhas (O i ) Cada coluna representa uma família de genes homólogos g1 g2 g3 g4 O1 O2 O3 O4 O5 O6
150 orthomcl pipeline Li Li et al. Genome Res. 2003; 13:
151
152
153
154 Fig. 4. Net gene loss or gain throughout the evolution of the {alpha}-proteobacterial species Boussau, Bastien et al. (2004) Proc. Natl. Acad. Sci. USA 101, September 2016 JC Setubal 154 Copyright 2004 by the National Academy of Sciences
 KEGG")
155 Protein family resources Clusters of orthologous groups (COG, KOG, eggnog) KEGG orthologs

156

157 Query by accession

158
159
160

161
162

163 Phylofacts query by sequence search

164 Resource federation: InterPro
165 Not as easy as it may sound Specific protein families may not be consistent across resources Most families (MSAs, trees, HMMs) in these resources are not manually curated Domains in Pfam-A are curated TIGRfams are curated HAMAP families are curated
166 Pan genoma; genoma core e genoma acessório A B core C pan: A U B U C acessório: pan - core 22 September 2016 JC Setubal 166
167 Curva de pan-genoma (n = 4)
168 Curva de core genoma (n = 4) O número de genes para x=1 não é o mesmo do gráfico anterior pois os singletons são descartados
169 Genomas fechados e abertos Fechado O número de genes/famílias do pan-genoma atinge um platô Aberto O número de genes/famílias não atinge um platô; cresce sempre que se acrescentam novos genomas
170 O genoma de E. coli é aberto Fonte: pangp.big.ac.cn
171 Conjuntos + contexto Como genes compartilhados aparecem em seus respectivos genomas? Filogenômica Busca de sintenia = preservação de ordem Basta fazer um alinhamento Os caracteres a serem alinhados são os genes
172 172

173 Proteome alignment done with LCS (top: Xcc; bottom: Xac ) Blue: BBHs that are in the LCS; dark blue: BBHs not in the LCS; red: Xac specifics; yellow: Xcc specifics 22 September 2016 JC Setubal 173

174 Roda da ortologia

175
176 Sumário - 1 Comparação de sequências curtas 2-a-2 Alinhamento Sistemas de pontuação Matrizes de substituição Programação dinâmica Significância estatística de alinhamentos BLAST
177 Sumário - 2 Comparação de várias sequências ao mesmo tempo Alinhamento múltiplo Programas Comparação de sequências longas 2-a-2 Alinhamento múltiplo Comparação entre conjuntos de genes

178

179

180 proteômica
Métodos de alinhamento de sequências biológicas. Marcelo Falsarella Carazzolle
 Métodos de alinhamento de sequências biológicas Marcelo Falsarella Carazzolle Resumo - Introdução - Alinhamentos ótimos - Global - Local (Smith-Waterman) - Semi global - Matrizes de alinhamento (BLOSUM)
Métodos de alinhamento de sequências biológicas Marcelo Falsarella Carazzolle Resumo - Introdução - Alinhamentos ótimos - Global - Local (Smith-Waterman) - Semi global - Matrizes de alinhamento (BLOSUM)
Alinhamento local- Utilização do BLAST
 Alinhamento local- Utilização do BLAST BLAST Tipos de BLAST (blastn) Compara nucleotídeos (blastp) Compara proteínas Utiliza nucleotídeo como query, este é traduzido nos seus 6 quadros de leitura e é comparado
Alinhamento local- Utilização do BLAST BLAST Tipos de BLAST (blastn) Compara nucleotídeos (blastp) Compara proteínas Utiliza nucleotídeo como query, este é traduzido nos seus 6 quadros de leitura e é comparado
alinhamento global-alinhamento múltiplo de seqüências
 alinhamento global-alinhamento múltiplo de seqüências Alinhamento múltiplos de seqüências Qual a importância de se realizar alinhamentos múltiplos em oposição a alinhamentos em pares? Alinhamento múltiplos
alinhamento global-alinhamento múltiplo de seqüências Alinhamento múltiplos de seqüências Qual a importância de se realizar alinhamentos múltiplos em oposição a alinhamentos em pares? Alinhamento múltiplos
PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE
 CONCEITOS EM EPIDEMIOLOGIA E FILOGENIA MOLECULARES PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE DE
CONCEITOS EM EPIDEMIOLOGIA E FILOGENIA MOLECULARES PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE DE
Algoritmos 3/17/ Algoritmos como área de estudo e investigação
 Algoritmos e Complexidade Ana Teresa Freitas INESC-ID/IST ID/IST 3/17/2005 1 O que é um algoritmo? Algoritmos: Sequência de instruções necessárias para a resolução de um problema bem formulado [passíveis
Algoritmos e Complexidade Ana Teresa Freitas INESC-ID/IST ID/IST 3/17/2005 1 O que é um algoritmo? Algoritmos: Sequência de instruções necessárias para a resolução de um problema bem formulado [passíveis
A matemática e o genoma. Resumo
 I Coloquio Regional da Região Centro-Oeste, 3 a 6 de novembro de 2009 Universidade Federal de Mato Grosso do Sul Mini-curso A matemática e o genoma Nalvo F. Almeida Jr. Resumo Os avanços da biotecnologia
I Coloquio Regional da Região Centro-Oeste, 3 a 6 de novembro de 2009 Universidade Federal de Mato Grosso do Sul Mini-curso A matemática e o genoma Nalvo F. Almeida Jr. Resumo Os avanços da biotecnologia
Anotação de Genomas. Fabiana G. S. Pinto
 Anotação de Genomas Fabiana G. S. Pinto Obtenção de Seqüências geradas pelo MegaBace 1000 Dados brutos (medidas analógicas) de saída do seqüênciamento Base calling BIOINFORMÁTICA * PHRED: - Transforma
Anotação de Genomas Fabiana G. S. Pinto Obtenção de Seqüências geradas pelo MegaBace 1000 Dados brutos (medidas analógicas) de saída do seqüênciamento Base calling BIOINFORMÁTICA * PHRED: - Transforma
Árvore Binária de Busca Ótima
 MAC 5710 - Estruturas de Dados - 2008 Referência bibliográfica Os slides sobre este assunto são parcialmente baseados nas seções sobre árvore binária de busca ótima do capítulo 4 do livro N. Wirth. Algorithms
MAC 5710 - Estruturas de Dados - 2008 Referência bibliográfica Os slides sobre este assunto são parcialmente baseados nas seções sobre árvore binária de busca ótima do capítulo 4 do livro N. Wirth. Algorithms
Comparação e alinhamento de sequências
 Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Explorando bancos de dados genômicos e introdução à bioinformática. Guilherme Targino Valente Marcos Tadeu Geraldo. Bioinformática
 Explorando bancos de dados genômicos e introdução à bioinformática Guilherme Targino Valente Marcos Tadeu Geraldo 22/07/2011 Bioinformática É a aplicação de estatística e ciência da computação no campo
Explorando bancos de dados genômicos e introdução à bioinformática Guilherme Targino Valente Marcos Tadeu Geraldo 22/07/2011 Bioinformática É a aplicação de estatística e ciência da computação no campo
DEFINIÇÕES EM EPIDEMIOLOGIA MOLECULAR E CONCEITOS BÁSICOS EM BIOLOGIA MOLECULAR
 DEFINIÇÕES EM E DEFINIÇÕES EM E CONCEITOS BÁSICOS EM BIOLOGIA PARA QUE SERVE ESTA AULA 1. DEFINIÇÕES EM CONCEITUAÇÃO DE DIFERENCIAÇÃO ENTRE, TAXONOMIA E FILOGENIA 2. CONCEITOS EM BIOLOGIA APRESENTAR (REVER)
DEFINIÇÕES EM E DEFINIÇÕES EM E CONCEITOS BÁSICOS EM BIOLOGIA PARA QUE SERVE ESTA AULA 1. DEFINIÇÕES EM CONCEITUAÇÃO DE DIFERENCIAÇÃO ENTRE, TAXONOMIA E FILOGENIA 2. CONCEITOS EM BIOLOGIA APRESENTAR (REVER)
BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011
 BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011 7ª aula teórica 11 Outubro 2010 Proteínas estruturais e funcionais Organização estrutural das proteínas Estrutura e diferentes funções de proteínas
BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011 7ª aula teórica 11 Outubro 2010 Proteínas estruturais e funcionais Organização estrutural das proteínas Estrutura e diferentes funções de proteínas
Bioinformática. Alinhamento de Sequências. Prof. Msc. Rommel Ramos
 Bioinformática Alinhamento de Sequências Prof. Msc. Rommel Ramos 2013 Sumário 1. Comparação de Sequências 2. O que é alinhamento? 3. Tipos de Alinhamento 4. Algoritmos 5. Métodos de Alinhamento Comparação
Bioinformática Alinhamento de Sequências Prof. Msc. Rommel Ramos 2013 Sumário 1. Comparação de Sequências 2. O que é alinhamento? 3. Tipos de Alinhamento 4. Algoritmos 5. Métodos de Alinhamento Comparação
Dezembro - 2006. Bioinformática. e Anotação. Eduardo Fernandes Formighieri Laboratório de Genômica e Expressão / UNICAMP
 Dezembro - 2006 Bioinformática e Anotação Eduardo Fernandes Formighieri Laboratório de Genômica e Expressão / UNICAMP Hoje 1. Introdução à Genômica 2. Introdução à Bioinformática 3. Introdução à Anotação
Dezembro - 2006 Bioinformática e Anotação Eduardo Fernandes Formighieri Laboratório de Genômica e Expressão / UNICAMP Hoje 1. Introdução à Genômica 2. Introdução à Bioinformática 3. Introdução à Anotação
IFSC Campus Lages. Tradução. Biologia Molecular Prof. Silmar Primieri
 IFSC Campus Lages Tradução Biologia Molecular Prof. Silmar Primieri Relação DNA RNA Proteína Estrutura das proteínas Gene - Proteína Hipótese Gene - Proteina Os genes são responsáveis pelo funcionamento
IFSC Campus Lages Tradução Biologia Molecular Prof. Silmar Primieri Relação DNA RNA Proteína Estrutura das proteínas Gene - Proteína Hipótese Gene - Proteina Os genes são responsáveis pelo funcionamento
Bioinformática e Genética Animal. Pâmela A. Alexandre Doutoranda
 Bioinformática e Genética Animal Pâmela A. Alexandre Doutoranda Descoberta da estrutura do DNA» Watson e Crick, 1953 DNA RNA Proteína Projeto Genoma Humano» 1990» 18 países» US$ 2,7 Bi» 13 anos (previsão
Bioinformática e Genética Animal Pâmela A. Alexandre Doutoranda Descoberta da estrutura do DNA» Watson e Crick, 1953 DNA RNA Proteína Projeto Genoma Humano» 1990» 18 países» US$ 2,7 Bi» 13 anos (previsão
CURSO: ENFERMAGEM DISCIPLINA: BIOQUÍMICA HUMANA PROF. WILLAME BEZERRA. Aminoácidos. Prof. Willame Bezerra
 CURSO: ENFERMAGEM DISCIPLINA: BIOQUÍMICA HUMANA PROF. WILLAME BEZERRA Aminoácidos Prof. Willame Bezerra As proteínas são as biomoléculas mais abundantes nos seres vivos e exercem funções fundamentais em
CURSO: ENFERMAGEM DISCIPLINA: BIOQUÍMICA HUMANA PROF. WILLAME BEZERRA Aminoácidos Prof. Willame Bezerra As proteínas são as biomoléculas mais abundantes nos seres vivos e exercem funções fundamentais em
Complexidade de Algoritmos
 Complexidade de Algoritmos! Uma característica importante de qualquer algoritmo é seu tempo de execução! é possível determiná-lo através de métodos empíricos, considerando-se entradas diversas! é também
Complexidade de Algoritmos! Uma característica importante de qualquer algoritmo é seu tempo de execução! é possível determiná-lo através de métodos empíricos, considerando-se entradas diversas! é também
BC1424 Algoritmos e Estruturas de Dados I Aula 05 Custos de um algoritmo e funções de complexidade
 BC1424 Algoritmos e Estruturas de Dados I Aula 05 Custos de um algoritmo e funções de complexidade Prof. Jesús P. Mena-Chalco 1Q-2016 1 1995 2015 2 Custo de um algoritmo e funções de complexidade Introdução
BC1424 Algoritmos e Estruturas de Dados I Aula 05 Custos de um algoritmo e funções de complexidade Prof. Jesús P. Mena-Chalco 1Q-2016 1 1995 2015 2 Custo de um algoritmo e funções de complexidade Introdução
Organização e Arquitetura de Computadores I
 Organização e Arquitetura de Computadores I Slide 1 Memória Virtual os primeiros computadores (início dos anos 60) tinham memória principal muito reduzida O PDP-1 funcionava com uma memória de 4096 palavras
Organização e Arquitetura de Computadores I Slide 1 Memória Virtual os primeiros computadores (início dos anos 60) tinham memória principal muito reduzida O PDP-1 funcionava com uma memória de 4096 palavras
Teoria e Prática de Sistemática Filogenética
 Disciplina BOT-99 PPG-BOT-INPA 2015 Teoria e Prática de Sistemática Filogenética Alberto Vicentini alberto.vicentini@inpa.gov.br Mário Henrique Terra Araujo araujo.mht@gmail.com Programa de Pós-Graduação
Disciplina BOT-99 PPG-BOT-INPA 2015 Teoria e Prática de Sistemática Filogenética Alberto Vicentini alberto.vicentini@inpa.gov.br Mário Henrique Terra Araujo araujo.mht@gmail.com Programa de Pós-Graduação
Resumo - capítulo 3 - Alinhamento de pares de sequências
 Resumo - capítulo 3 - Alinhamento de pares de sequências Pedro Ivo Gomes de Faria Sumário 1 Introdução 3 1.1 Definição de alinhamento de sequências............. 3 1.1.1 Alinhamento global....................
Resumo - capítulo 3 - Alinhamento de pares de sequências Pedro Ivo Gomes de Faria Sumário 1 Introdução 3 1.1 Definição de alinhamento de sequências............. 3 1.1.1 Alinhamento global....................
Universidade Estadual de Maringá - UEM
 Universidade Estadual de Maringá - UEM Disciplina: Biologia Molecular 6855 T1 e T2 Ciências Biológicas Transcriptoma metodologia ORESTES Profa. Dra. Maria Aparecida Fernandez Estratégia ORESTES ESTs de
Universidade Estadual de Maringá - UEM Disciplina: Biologia Molecular 6855 T1 e T2 Ciências Biológicas Transcriptoma metodologia ORESTES Profa. Dra. Maria Aparecida Fernandez Estratégia ORESTES ESTs de
Aspectos de genômica comparativa
 Aspectos de genômica comparativa Carlos Juliano Moura Viana Dissertação de Mestrado Orientação:Prof.Dr. Nalvo Franco de Almeida Jr. Área de Concentração: Biologia Computacional Durante a elaboração desse
Aspectos de genômica comparativa Carlos Juliano Moura Viana Dissertação de Mestrado Orientação:Prof.Dr. Nalvo Franco de Almeida Jr. Área de Concentração: Biologia Computacional Durante a elaboração desse
Motantagem de Contigs de sequências de genomas e Transcriptomas. Introdução
 Motantagem de Contigs de sequências de genomas e Transcriptomas Introdução As novas tecnologias de sequenciamento conseguem produzir uma quantidade de dados muito grande com custos baixos. A velocidade
Motantagem de Contigs de sequências de genomas e Transcriptomas Introdução As novas tecnologias de sequenciamento conseguem produzir uma quantidade de dados muito grande com custos baixos. A velocidade
Compiladores - Análise Ascendente
 Compiladores - Análise Ascendente Fabio Mascarenhas - 2013.1 http://www.dcc.ufrj.br/~fabiom/comp Análise Descendente vs. Ascendente As técnicas de análise que vimos até agora (recursiva com retrocesso,
Compiladores - Análise Ascendente Fabio Mascarenhas - 2013.1 http://www.dcc.ufrj.br/~fabiom/comp Análise Descendente vs. Ascendente As técnicas de análise que vimos até agora (recursiva com retrocesso,
Metagenômica e sequenciamento de nova geração. Fabrício Campos 25 de junho de 2015
 Metagenômica e sequenciamento de nova geração Fabrício Campos 25 de junho de 2015 Conceitos METAGENOMA É o genoma coletivo do microbioma total encontrado em um determinado habitat METAGENÔMICA É a análise
Metagenômica e sequenciamento de nova geração Fabrício Campos 25 de junho de 2015 Conceitos METAGENOMA É o genoma coletivo do microbioma total encontrado em um determinado habitat METAGENÔMICA É a análise
Técnicas de Programação III Análise de Algoritmos (Continuação)
") Técnicas de Programação III Análise de Algoritmos (Continuação) Aula ministrada em: 23/08/2007 Prof. Mauro L. C. Silva 1/10 Objetivos da Aula Entender a Análise e a Complexidade de Algoritmos 2/10 Avaliação
Técnicas de Programação III Análise de Algoritmos (Continuação) Aula ministrada em: 23/08/2007 Prof. Mauro L. C. Silva 1/10 Objetivos da Aula Entender a Análise e a Complexidade de Algoritmos 2/10 Avaliação
COLÉGIO PEDRO II CAMPUS TIJUCA II. DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI
 COLÉGIO PEDRO II CAMPUS TIJUCA II DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI 1º & 2º TURNOS 3ª SÉRIE / ENSINO MÉDIO REGULAR & INTEGRADO ANO LETIVO 2015 PROFESSORES: FRED & PEDRO
COLÉGIO PEDRO II CAMPUS TIJUCA II DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI 1º & 2º TURNOS 3ª SÉRIE / ENSINO MÉDIO REGULAR & INTEGRADO ANO LETIVO 2015 PROFESSORES: FRED & PEDRO
Estruturas de Dados Pilhas, Filas, Listas
 PMR2300 Escola Politécnica da Universidade de São Paulo Introdução Estruturas de dados são objetos que armazenam dados de forma eficiente, oferecendo certos serviços para o usuário (ordenação eficiente
PMR2300 Escola Politécnica da Universidade de São Paulo Introdução Estruturas de dados são objetos que armazenam dados de forma eficiente, oferecendo certos serviços para o usuário (ordenação eficiente
Caracterização e classificação de proteínas. Desafio de Paracelsus
 Objetivos FFI0776 Modelagem e Engenharia de Proteínas Prof. Rafael V. C. Guido rvcguido@ifsc.usp.br Aula 05 Caracterização e classificação de proteínas Proteínas homólogas Base de dados SCOP CATH Desafio
Objetivos FFI0776 Modelagem e Engenharia de Proteínas Prof. Rafael V. C. Guido rvcguido@ifsc.usp.br Aula 05 Caracterização e classificação de proteínas Proteínas homólogas Base de dados SCOP CATH Desafio
DCBD. Avaliação de modelos. Métricas para avaliação de desempenho. Avaliação de modelos. Métricas para avaliação de desempenho...
 DCBD Métricas para avaliação de desempenho Como avaliar o desempenho de um modelo? Métodos para avaliação de desempenho Como obter estimativas confiáveis? Métodos para comparação de modelos Como comparar
DCBD Métricas para avaliação de desempenho Como avaliar o desempenho de um modelo? Métodos para avaliação de desempenho Como obter estimativas confiáveis? Métodos para comparação de modelos Como comparar
Algoritmos Avançados Análise de Complexidade
 CCAE Centro de Ciências Aplicadas e Educação UFPB - Campus IV - Litoral Norte Algoritmos Avançados Análise de Complexidade COMPLEXIDADE DE ALGORITMOS Definição: A Complexidade de um Algoritmo consiste
CCAE Centro de Ciências Aplicadas e Educação UFPB - Campus IV - Litoral Norte Algoritmos Avançados Análise de Complexidade COMPLEXIDADE DE ALGORITMOS Definição: A Complexidade de um Algoritmo consiste
Exemplos de Aplicações da Teoria das Probabilidades em Biologia. Qual a probabilidade de que o próximo nucleotídeo na seqüência seja A, C, G ou T?
 Exemplos de Aplicações da Teoria das Probabilidades em Biologia Exemplo 1. Suponha que se conheça a seguinte seqüência de nucleotídeos em uma molécula de DNA: AGCTTCCGATCCGCTATAATCGTTAGTTGTTACACCTCTG Qual
Exemplos de Aplicações da Teoria das Probabilidades em Biologia Exemplo 1. Suponha que se conheça a seguinte seqüência de nucleotídeos em uma molécula de DNA: AGCTTCCGATCCGCTATAATCGTTAGTTGTTACACCTCTG Qual
Inteligência Artificial
 Inteligência Artificial Aula 6 Algoritmos Genéticos M.e Guylerme Velasco Roteiro Introdução Otimização Algoritmos Genéticos Representação Seleção Operadores Geneticos Aplicação Caixeiro Viajante Introdução
Inteligência Artificial Aula 6 Algoritmos Genéticos M.e Guylerme Velasco Roteiro Introdução Otimização Algoritmos Genéticos Representação Seleção Operadores Geneticos Aplicação Caixeiro Viajante Introdução
Área que visa determinar a complexidade (custo) de um algoritmo, com isso é possível:
 de um algoritmo, com isso é possível:") Área que visa determinar a complexidade (custo) de um algoritmo, com isso é possível: Comparar algoritmos: existem algoritmos que resolvem o mesmo tipo de problema. Determinar se o algoritmo é ótimo :
Área que visa determinar a complexidade (custo) de um algoritmo, com isso é possível: Comparar algoritmos: existem algoritmos que resolvem o mesmo tipo de problema. Determinar se o algoritmo é ótimo :
IACB 1º Semestre de 2014/2015. Exercicios de Preparação para o Teste 1
 IACB 1º Semestre de 2014/2015 Exercicios de Preparação para o Teste 1 Introdução (0 ou 1 questão no teste 1) 1. O que é a BioInformática? Resposta: Bioinformática é um campo interdisciplinar que aplica
IACB 1º Semestre de 2014/2015 Exercicios de Preparação para o Teste 1 Introdução (0 ou 1 questão no teste 1) 1. O que é a BioInformática? Resposta: Bioinformática é um campo interdisciplinar que aplica
Bioinformática. João Varela Aula T7
 Bioinformática C U R S O S E M B I O L O G I A, B I O Q U Í M I C A, B I O T E C N O L O G I A, C I Ê N C I A S B I O M É D I C A S E E N G E N H A R I A B I O L Ó G I C A João Varela jvarela@ualg.pt Aula
Bioinformática C U R S O S E M B I O L O G I A, B I O Q U Í M I C A, B I O T E C N O L O G I A, C I Ê N C I A S B I O M É D I C A S E E N G E N H A R I A B I O L Ó G I C A João Varela jvarela@ualg.pt Aula
Soluções de Conjunto de Problemas 1
 Soluções de 7.012 Conjunto de Problemas 1 Questão 1 a) Quais são os quatro tipos principais de moléculas biológicas discutidos na aula? Cite uma função importante de cada tipo de molécula biológica na
Soluções de 7.012 Conjunto de Problemas 1 Questão 1 a) Quais são os quatro tipos principais de moléculas biológicas discutidos na aula? Cite uma função importante de cada tipo de molécula biológica na
ANÁLISE DE ALGORITMOS: PARTE 3
 ANÁLISE DE ALGORITMOS: PARTE 3 Prof. André Backes 2 A notação grande-o é a forma mais conhecida e utilizada de análise Complexidade do nosso algoritmo no pior caso Seja de tempo ou de espaço É o caso mais
ANÁLISE DE ALGORITMOS: PARTE 3 Prof. André Backes 2 A notação grande-o é a forma mais conhecida e utilizada de análise Complexidade do nosso algoritmo no pior caso Seja de tempo ou de espaço É o caso mais
Algoritmos para Genómica Comparativa
 Universidade de Aveiro 2010 Departamento Electrónica, Telecomunicações e Informática Vasco da Rocha Figueiras Algoritmos para Genómica Comparativa Universidade de Aveiro 2010 Departamento de Electrónica,
Universidade de Aveiro 2010 Departamento Electrónica, Telecomunicações e Informática Vasco da Rocha Figueiras Algoritmos para Genómica Comparativa Universidade de Aveiro 2010 Departamento de Electrónica,
Apostila 01 Fundamentação da Teoria da Computação e Linguagens Formais
 Cursos: Bacharelado em Ciência da Computação e Bacharelado em Sistemas de Informação Disciplinas: (1493A) Teoria da Computação e Linguagens Formais, (4623A) Teoria da Computação e Linguagens Formais e
Cursos: Bacharelado em Ciência da Computação e Bacharelado em Sistemas de Informação Disciplinas: (1493A) Teoria da Computação e Linguagens Formais, (4623A) Teoria da Computação e Linguagens Formais e
Capítulo 6. Alinhamentos múltiplos de sequências macromoleculares.
 Capítulo 6. Alinhamentos múltiplos de sequências macromoleculares. versão 0.5 Como vimos no capítulo anterior, o procedimento de alinhamento entre sequências de macromoléculas equivale ao estabelecimento
Capítulo 6. Alinhamentos múltiplos de sequências macromoleculares. versão 0.5 Como vimos no capítulo anterior, o procedimento de alinhamento entre sequências de macromoléculas equivale ao estabelecimento
Complexidade de Algoritmos
 60 Desempenho 50 40 30 20 Algoritmo1 Algoritmo2 Algoritmo3 10 0 Complexidade de Algoritmos INFORMÁTICA BÁSICA Prof. Demétrios Coutinho C a m p u s P a u d o s F e r r o s D i s c i p l i n a d e A l g
60 Desempenho 50 40 30 20 Algoritmo1 Algoritmo2 Algoritmo3 10 0 Complexidade de Algoritmos INFORMÁTICA BÁSICA Prof. Demétrios Coutinho C a m p u s P a u d o s F e r r o s D i s c i p l i n a d e A l g
Aula 7: Portas Lógicas: AND, OR, NOT, XOR, NAND e NOR
 Aula 7: Portas Lógicas: AND, OR, NOT, XOR, NAND e NOR Conforme discutido na última aula, cada operação lógica possui sua própria tabela verdade. A seguir será apresentado o conjunto básico de portas lógicas
Aula 7: Portas Lógicas: AND, OR, NOT, XOR, NAND e NOR Conforme discutido na última aula, cada operação lógica possui sua própria tabela verdade. A seguir será apresentado o conjunto básico de portas lógicas
Complemento a Um e Complemento a Dois
 Complemento a Um e Complemento a Dois Cristina Boeres (baseado no material de Fernanda Passos) Instituto de Computação (UFF) Fundamentos de Arquiteturas de Computadores Cristina Boeres (IC/UFF) Complemento
Complemento a Um e Complemento a Dois Cristina Boeres (baseado no material de Fernanda Passos) Instituto de Computação (UFF) Fundamentos de Arquiteturas de Computadores Cristina Boeres (IC/UFF) Complemento
Algoritmos Paralelos Eficientes para Alguns Problemas em Proce. Caracteres
 lgoritmos Paralelos Eficientes para lguns Problemas em Processamento de Cadeias de Caracteres JI/SBC 2007 - Rio de Janeiro Parte 1 gradecimentos O material deste curso é baseado em trabalhos publicados
lgoritmos Paralelos Eficientes para lguns Problemas em Processamento de Cadeias de Caracteres JI/SBC 2007 - Rio de Janeiro Parte 1 gradecimentos O material deste curso é baseado em trabalhos publicados
COMPARAÇÃO DE SEQÜÊNCIAS DE DNA
 144 COMPARAÇÃO DE SEQÜÊNCIAS DE DNA PUCCI NETO, João 1 Resumo: A comparação de seqüências é uma operação básica muito importante na área de biologia computacional. Neste trabalho, é implementado um algoritmo
144 COMPARAÇÃO DE SEQÜÊNCIAS DE DNA PUCCI NETO, João 1 Resumo: A comparação de seqüências é uma operação básica muito importante na área de biologia computacional. Neste trabalho, é implementado um algoritmo
Soluções para Conjunto de Problemas 1
 Soluções para 7.012 Conjunto de Problemas 1 Pergunta 1 a) Quais são os quatro principais tipos de moléculas biológicas discutidas na palestra? Cite uma função importante para cada tipo de molécula biológica
Soluções para 7.012 Conjunto de Problemas 1 Pergunta 1 a) Quais são os quatro principais tipos de moléculas biológicas discutidas na palestra? Cite uma função importante para cada tipo de molécula biológica
Introdução à Programação
 Introdução à Program João Manuel R. S. Tavares Sumário 1. Ciclo de desenvolvimento de um programa; 2. Descrição de algoritmos; 3. Desenvolvimento modular de programas; 4. Estruturas de controlo de um programa.
Introdução à Program João Manuel R. S. Tavares Sumário 1. Ciclo de desenvolvimento de um programa; 2. Descrição de algoritmos; 3. Desenvolvimento modular de programas; 4. Estruturas de controlo de um programa.
Experimentos e Resultados
 6 Experimentos e Resultados Neste capítulo apresentamos os experimentos realizados e os resultados obtidos pelas heurísticas apresentadas. A primeira parte do capítulo aborda os experimentos e resultados
6 Experimentos e Resultados Neste capítulo apresentamos os experimentos realizados e os resultados obtidos pelas heurísticas apresentadas. A primeira parte do capítulo aborda os experimentos e resultados
Estruturas de Dados 2
 Estruturas de Dados 2 Análise Empírica de Algoritmos IF64C Estruturas de Dados 2 Engenharia da Computação Prof. João Alberto Fabro - Slide 1/13 Análise da Eficiência de Algoritmos: Velocidade de Execução;
Estruturas de Dados 2 Análise Empírica de Algoritmos IF64C Estruturas de Dados 2 Engenharia da Computação Prof. João Alberto Fabro - Slide 1/13 Análise da Eficiência de Algoritmos: Velocidade de Execução;
Análise e Projeto de Algoritmos
 Análise e Projeto de Algoritmos Mestrado em Ciência da Computação Prof. Dr. Aparecido Nilceu Marana Faculdade de Ciências I think the design of efficient algorithms is somehow the core of computer science.
Análise e Projeto de Algoritmos Mestrado em Ciência da Computação Prof. Dr. Aparecido Nilceu Marana Faculdade de Ciências I think the design of efficient algorithms is somehow the core of computer science.
Disciplina Evolução Módulo II. Prof. Carolina Voloch. Filogenia
 Disciplina Evolução Módulo II Prof. arolina Voloch Filogenia A sistemática é a ciência que une a taxonomia, ou seja, a ciência da classificação dos organismos, com a filogenia, a ciência que traça a história
Disciplina Evolução Módulo II Prof. arolina Voloch Filogenia A sistemática é a ciência que une a taxonomia, ou seja, a ciência da classificação dos organismos, com a filogenia, a ciência que traça a história
LÓGICA I. André Pontes
 LÓGICA I André Pontes 1. Conceitos fundamentais O que é a Lógica? A LÓGICA ENQUANTO DISCIPLINA Estudo das leis de preservação da verdade. [Frege; O Pensamento] Estudo das formas válidas de argumentos.
LÓGICA I André Pontes 1. Conceitos fundamentais O que é a Lógica? A LÓGICA ENQUANTO DISCIPLINA Estudo das leis de preservação da verdade. [Frege; O Pensamento] Estudo das formas válidas de argumentos.
Árvores Filogenéticas
 Árvores Filogenéticas 1 Introdução todos os fundamentos da biologia moderna estão associados à teoria da evolução de Darwin. de aspectos de anatomia, passando por comportamento e chegando à genética, toda
Árvores Filogenéticas 1 Introdução todos os fundamentos da biologia moderna estão associados à teoria da evolução de Darwin. de aspectos de anatomia, passando por comportamento e chegando à genética, toda
Nome da atividade: Identificação de uma proteína a partir da sua seqüência nucleotídica e determinação da sua estrutura e função
 Caros Alunos, Para desenvolver a atividade abaixo vocês precisarão de um computador conectado à internet banda larga, do arquivo.txt (bloco de notas) disponível no Constructore e deste arquivo com as coordenadas
Caros Alunos, Para desenvolver a atividade abaixo vocês precisarão de um computador conectado à internet banda larga, do arquivo.txt (bloco de notas) disponível no Constructore e deste arquivo com as coordenadas
Bioinformática. João Varela jvarela@ualg.pt. Aula T4 CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, CIÊNCIAS BIOMÉDICAS E ENGENHARIA BIOLÓGICA
 Bioinformática CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, CIÊNCIAS BIOMÉDICAS E ENGENHARIA BIOLÓGICA João Varela jvarela@ualg.pt Aula T4 Esquema de anotação Annothaton 1. Determinar a localização das
Bioinformática CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, CIÊNCIAS BIOMÉDICAS E ENGENHARIA BIOLÓGICA João Varela jvarela@ualg.pt Aula T4 Esquema de anotação Annothaton 1. Determinar a localização das
UFABC Bacharelado em Ciência & Tecnologia
 UFABC Bacharelado em Ciência & Tecnologia Transformações Bioquímicas (BC0308) Prof Luciano Puzer http://professor.ufabc.edu.br/~luciano.puzer/ Propriedades, funções e transformações de aminoácidos e proteínas
UFABC Bacharelado em Ciência & Tecnologia Transformações Bioquímicas (BC0308) Prof Luciano Puzer http://professor.ufabc.edu.br/~luciano.puzer/ Propriedades, funções e transformações de aminoácidos e proteínas
Correção Ortográfica. Processamento Estatístico da Linguagem Natural. Correção de Erros. Distância Mínima de Edição. Distância Mínima de Edição
 Processamento Estatístico da Linguagem Natural Aula 6 Professora Bianca (Sala 302 Bloco E) bianca@ic.uff.br http://www.ic.uff.br/~bianca/peln/ Correção Ortográfica Três tipos de problemas: Detecção de
Processamento Estatístico da Linguagem Natural Aula 6 Professora Bianca (Sala 302 Bloco E) bianca@ic.uff.br http://www.ic.uff.br/~bianca/peln/ Correção Ortográfica Três tipos de problemas: Detecção de
Documentos. ISSN 0102-0110 Dezembro, 2007 224. O programa BLAST: guia prático de utilização
 Documentos ISSN 0102-0110 Dezembro, 2007 224 X O programa BLAST: guia prático de utilização ISSN 0102 0110 Dezembro, 2007 Empresa Brasileira de Pesquisa Agropecuária Embrapa Recursos Genéticos e Biotecnologia
Documentos ISSN 0102-0110 Dezembro, 2007 224 X O programa BLAST: guia prático de utilização ISSN 0102 0110 Dezembro, 2007 Empresa Brasileira de Pesquisa Agropecuária Embrapa Recursos Genéticos e Biotecnologia
Princípios de Sistemática Molecular
 ! Ciências teóricas e sistemática biológica "! DNA, genes, código genético e mutação! Alinhamento de seqüências! Mudanças evolutivas em seqüências de nucleotídeos! Otimização em espaços contínuos e discretos!
! Ciências teóricas e sistemática biológica "! DNA, genes, código genético e mutação! Alinhamento de seqüências! Mudanças evolutivas em seqüências de nucleotídeos! Otimização em espaços contínuos e discretos!
Ecologia e Modelagem Ambiental para a conservação da Biodiversidade
 Ecologia e Modelagem Ambiental para a conservação da Biodiversidade SISTEMÁTICA FILOGENÉTICA, BOTÂNICA E CONSERVAÇÃO 1. NOÇÕES BÁSICAS DE SISTEMÁTICA FILOGENÉTICA 2. Índice de Diversidade Filogenética
Ecologia e Modelagem Ambiental para a conservação da Biodiversidade SISTEMÁTICA FILOGENÉTICA, BOTÂNICA E CONSERVAÇÃO 1. NOÇÕES BÁSICAS DE SISTEMÁTICA FILOGENÉTICA 2. Índice de Diversidade Filogenética
Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática
 Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática Hoje iremos trabalhar com dois programas free desenvolvidos pelo Sanger institute: Artemis e ACT. Artemis
Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática Hoje iremos trabalhar com dois programas free desenvolvidos pelo Sanger institute: Artemis e ACT. Artemis
5. Expressões aritméticas
 5. Expressões aritméticas 5.1. Conceito de Expressão O conceito de expressão em termos computacionais está intimamente ligado ao conceito de expressão (ou fórmula) matemática, onde um conjunto de variáveis
5. Expressões aritméticas 5.1. Conceito de Expressão O conceito de expressão em termos computacionais está intimamente ligado ao conceito de expressão (ou fórmula) matemática, onde um conjunto de variáveis
IME, UFF 7 de novembro de 2013
 em Lógica IME, UFF 7 de novembro de 2013 em Sumário Intermezzo sobre problemas. Intermezzo sobre algoritmos.. em : Val, Sat, Conseq, Equiv, Consist. Redução de problemas. em Um problema computacional é
em Lógica IME, UFF 7 de novembro de 2013 em Sumário Intermezzo sobre problemas. Intermezzo sobre algoritmos.. em : Val, Sat, Conseq, Equiv, Consist. Redução de problemas. em Um problema computacional é
Computação paralela para problemas em Biologia
 Computação paralela para problemas em Biologia ERAD-SP - julho de 2013 - São Carlos Computação paralela: hoje e amanhã e desafios. Bioinformática: área interdisciplinar onde há uma simbiose de duas importantes
Computação paralela para problemas em Biologia ERAD-SP - julho de 2013 - São Carlos Computação paralela: hoje e amanhã e desafios. Bioinformática: área interdisciplinar onde há uma simbiose de duas importantes
Criando Cladogramas. Introdução. Objetivos: Construir e analisar um cladograma. Procedimento
 Criando Cladogramas Introdução Uma forma de descobrir o grau de parentesco de seres vivos (filogenia) é comparar as estruturas anatômicas de vários organismos diferentes. Órgãos e estruturas que são parecidos
Criando Cladogramas Introdução Uma forma de descobrir o grau de parentesco de seres vivos (filogenia) é comparar as estruturas anatômicas de vários organismos diferentes. Órgãos e estruturas que são parecidos
SEO e UX - 2016. www.infotechnology.com.br
 SEO e UX - 2016 www.infotechnology.com.br Objetivos UX SEO 2 SEO - Search engine optimazation SEO é um conjunto de técnicas que têm como objetivo principal tornar os sites amigáveis para os sites de busca
SEO e UX - 2016 www.infotechnology.com.br Objetivos UX SEO 2 SEO - Search engine optimazation SEO é um conjunto de técnicas que têm como objetivo principal tornar os sites amigáveis para os sites de busca
Computação na Biologia Molecular e Bionanotecnologia: Computação Biológica
 Computação na Biologia Molecular e Bionanotecnologia: Computação Biológica Leila Ribeiro Instituto de Informática -UFRGS Roteiro Minhas áreas de interesse... Evolução da Ciência da Computação Biologia
Computação na Biologia Molecular e Bionanotecnologia: Computação Biológica Leila Ribeiro Instituto de Informática -UFRGS Roteiro Minhas áreas de interesse... Evolução da Ciência da Computação Biologia
Tratamento dos Erros de Sintaxe. Adriano Maranhão
 Tratamento dos Erros de Sintaxe Adriano Maranhão Introdução Se um compilador tivesse que processar somente programas corretos, seu projeto e sua implementação seriam grandemente simplificados. Mas os programadores
Tratamento dos Erros de Sintaxe Adriano Maranhão Introdução Se um compilador tivesse que processar somente programas corretos, seu projeto e sua implementação seriam grandemente simplificados. Mas os programadores
Compiladores Analisador Sintático. Prof. Antonio Felicio Netto Ciência da Computação
 Compiladores Analisador Sintático Prof. Antonio Felicio Netto antonio.felicio@anhanguera.com Ciência da Computação 1 Análise Sintática - A Análise Sintática constitui a segunda fase de um tradutor de uma
Compiladores Analisador Sintático Prof. Antonio Felicio Netto antonio.felicio@anhanguera.com Ciência da Computação 1 Análise Sintática - A Análise Sintática constitui a segunda fase de um tradutor de uma
Roberto Geraldo Tavares Arnaut Gustavo de Figueiredo Tarcsay. Potenciação. Sanja Gjenero. Fonte:
 Potenciação 31 Sanja Gjenero Roberto Geraldo Tavares Arnaut Gustavo de Figueiredo Tarcsay Fonte: www.sxc.hu e-tec Brasil Estatística Aplicada META Apresentar as operações de potenciação. OBJETIVOS PRÉ-REQUISITOS
Potenciação 31 Sanja Gjenero Roberto Geraldo Tavares Arnaut Gustavo de Figueiredo Tarcsay Fonte: www.sxc.hu e-tec Brasil Estatística Aplicada META Apresentar as operações de potenciação. OBJETIVOS PRÉ-REQUISITOS
03 Análise de Algoritmos (parte 3) SCC201/501 - Introdução à Ciência de Computação II
 SCC201/501 - Introdução à Ciência de Computação II") 03 Análise de Algoritmos (parte 3) SCC201/501 - Introdução à Ciência de Computação II Prof. Moacir Ponti Jr. www.icmc.usp.br/~moacir Instituto de Ciências Matemáticas e de Computação USP 2010/2 Moacir
03 Análise de Algoritmos (parte 3) SCC201/501 - Introdução à Ciência de Computação II Prof. Moacir Ponti Jr. www.icmc.usp.br/~moacir Instituto de Ciências Matemáticas e de Computação USP 2010/2 Moacir
Quantidade de memória necessária
 Tempo de processamento Um algoritmo que realiza uma tarefa em 10 horas é melhor que outro que realiza em 10 dias Quantidade de memória necessária Um algoritmo que usa 1MB de memória RAM é melhor que outro
Tempo de processamento Um algoritmo que realiza uma tarefa em 10 horas é melhor que outro que realiza em 10 dias Quantidade de memória necessária Um algoritmo que usa 1MB de memória RAM é melhor que outro
PLANO ANALÍTICO DA DISCIPLINA DE ESTRUTURA DE DADOS E ALGORÍTMOS
 O Regente PLANO ANALÍTICO DA DISCIPLINA DE ESTRUTURA DE DADOS E ALGORÍTMOS Curso: Licenciatura em Informtica Ano: 2º Semestre: 3º Docente: Arlete Ferrão/Calisto Comé Monitor: Edson Pessane Ano Lectivo:
O Regente PLANO ANALÍTICO DA DISCIPLINA DE ESTRUTURA DE DADOS E ALGORÍTMOS Curso: Licenciatura em Informtica Ano: 2º Semestre: 3º Docente: Arlete Ferrão/Calisto Comé Monitor: Edson Pessane Ano Lectivo:
Universidade Federal de Campina Grande Departamento de Sistemas e Computação Curso de Bacharelado em Ciência da Computação.
 Universidade Federal de Campina Grande Departamento de Sistemas e Computação Curso de Bacharelado em Ciência da Computação Organização e Arquitetura de Computadores I Organização e Arquitetura Básicas
Universidade Federal de Campina Grande Departamento de Sistemas e Computação Curso de Bacharelado em Ciência da Computação Organização e Arquitetura de Computadores I Organização e Arquitetura Básicas
Lista de exercícios sobre contagem de operações Prof. João B. Oliveira
 Lista de exercícios sobre contagem de operações Prof. João B. Oliveira 1. metodo m ( Vetor V ) int i, res = 0; para i de 1 a V.size res = res + V[i]; return res; Soma de elementos de um vetor, O( ). 2.
Lista de exercícios sobre contagem de operações Prof. João B. Oliveira 1. metodo m ( Vetor V ) int i, res = 0; para i de 1 a V.size res = res + V[i]; return res; Soma de elementos de um vetor, O( ). 2.
Linguagem C Princípios Básicos (parte 1)
") Linguagem C Princípios Básicos (parte 1) Objetivos O principal objetivo deste artigo é explicar alguns conceitos fundamentais de programação em C. No final será implementado um programa envolvendo todos
Linguagem C Princípios Básicos (parte 1) Objetivos O principal objetivo deste artigo é explicar alguns conceitos fundamentais de programação em C. No final será implementado um programa envolvendo todos
Como modelar o comportamento de um sistema? MAB-515
 Como modelar o comportamento de um sistema? MAB-515 Possibilidades de modelagem PARAMETRIZA modelo matemático experimento real AJUDA A COMPREENDER SIMULAÇÃO SOLUÇÃO ANALÍTICA MEDIDAS EXPERIMENTAIS NO MODELO
Como modelar o comportamento de um sistema? MAB-515 Possibilidades de modelagem PARAMETRIZA modelo matemático experimento real AJUDA A COMPREENDER SIMULAÇÃO SOLUÇÃO ANALÍTICA MEDIDAS EXPERIMENTAIS NO MODELO
Vamos estudar o que se entende por «programação», que é uma linguagem de programação e ver algumas terminologias própria de programação e como
 Vamos estudar o que se entende por «programação», que é uma linguagem de programação e ver algumas terminologias própria de programação e como utilizá-la. 1 Por si só, uma equipe não é muito inteligente.
Vamos estudar o que se entende por «programação», que é uma linguagem de programação e ver algumas terminologias própria de programação e como utilizá-la. 1 Por si só, uma equipe não é muito inteligente.
Introdução à Computação: Sistemas de Numeração
 Introdução à Computação: Sistemas de Numeração Beatriz F. M. Souza (bfmartins@inf.ufes.br) http://inf.ufes.br/~bfmartins/ Computer Science Department Federal University of Espírito Santo (Ufes), Vitória,
Introdução à Computação: Sistemas de Numeração Beatriz F. M. Souza (bfmartins@inf.ufes.br) http://inf.ufes.br/~bfmartins/ Computer Science Department Federal University of Espírito Santo (Ufes), Vitória,
1. Avaliação de impacto de programas sociais: por que, para que e quando fazer? (Cap. 1 do livro) 2. Estatística e Planilhas Eletrônicas 3.
 2. Estatística e Planilhas Eletrônicas 3.") 1 1. Avaliação de impacto de programas sociais: por que, para que e quando fazer? (Cap. 1 do livro) 2. Estatística e Planilhas Eletrônicas 3. Modelo de Resultados Potenciais e Aleatorização (Cap. 2 e 3
1 1. Avaliação de impacto de programas sociais: por que, para que e quando fazer? (Cap. 1 do livro) 2. Estatística e Planilhas Eletrônicas 3. Modelo de Resultados Potenciais e Aleatorização (Cap. 2 e 3
Instituto de Matemática e Estatística, UFF Março de 2011
 ,,,,, Instituto de Matemática e Estatística, UFF Março de 2011 ,, Sumário,,. finitos,. conjunto: por lista, por propriedade.. Igualdade,. Propriedades básicas.. ,, Christos Papadimitriou, Autor dos livros
,,,,, Instituto de Matemática e Estatística, UFF Março de 2011 ,, Sumário,,. finitos,. conjunto: por lista, por propriedade.. Igualdade,. Propriedades básicas.. ,, Christos Papadimitriou, Autor dos livros
2 Fluxogramas e Pseudocódigo
 2 Fluxogramas e Pseudocódigo Programação em C/C++ estrutura básica e conceitos fundamentais 1 Algoritmos em linguagem informal e em linguagens formais Voltemos a considerar alguns algoritmos que traduzem
2 Fluxogramas e Pseudocódigo Programação em C/C++ estrutura básica e conceitos fundamentais 1 Algoritmos em linguagem informal e em linguagens formais Voltemos a considerar alguns algoritmos que traduzem
Algoritmos Genéticos. Estéfane G. M. de Lacerda DCA/UFRN Outubro/2008
 Estéfane G. M. de Lacerda DCA/UFRN Outubro/2008 Introdução São técnicas de busca e otimização. É a metáfora da teoria da evolução das espécies iniciada pelo Fisiologista e Naturalista inglês Charles Darwin.
Estéfane G. M. de Lacerda DCA/UFRN Outubro/2008 Introdução São técnicas de busca e otimização. É a metáfora da teoria da evolução das espécies iniciada pelo Fisiologista e Naturalista inglês Charles Darwin.
Introdução a Programação
 Introdução a Programação Prof. André Gustavo Duarte de Almeida andre.almeida@ifrn.edu.br docente.ifrn.edu.br/andrealmeida Aula 02 Primeiro Programa Roteiro Primeiros Passos Variáveis Expressões Comandos
Introdução a Programação Prof. André Gustavo Duarte de Almeida andre.almeida@ifrn.edu.br docente.ifrn.edu.br/andrealmeida Aula 02 Primeiro Programa Roteiro Primeiros Passos Variáveis Expressões Comandos
Juliana Kaizer Vizzotto. Universidade Federal de Santa Maria. Disciplina de Teoria da Computação
 Universidade Federal de Santa Maria Disciplina de Teoria da Computação Quais são as capacidades e limitações fundamentais dos computadores? Funções Computáveis Algoritmo: descrição finitade uma computação
Universidade Federal de Santa Maria Disciplina de Teoria da Computação Quais são as capacidades e limitações fundamentais dos computadores? Funções Computáveis Algoritmo: descrição finitade uma computação
Hashing: conceitos. Hashing
 Hashing: conceitos hashing é uma técnica conhecida como espalhamento, mapeamento ou randomização que tenta distribuir dados em posições aleatórias de uma tabela (array) associa cada objeto (de um determinado
Hashing: conceitos hashing é uma técnica conhecida como espalhamento, mapeamento ou randomização que tenta distribuir dados em posições aleatórias de uma tabela (array) associa cada objeto (de um determinado
Introdução a Bioinformática
 Pontifícia Universidade Católica de Goiás Departamento de Biologia Disciplina: Bioinformática Bio1015 Introdução a Bioinformática Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com EMENTA Introdução
Pontifícia Universidade Católica de Goiás Departamento de Biologia Disciplina: Bioinformática Bio1015 Introdução a Bioinformática Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com EMENTA Introdução
Compiladores - Gramáticas
 Compiladores - Gramáticas Fabio Mascarenhas - 2013.1 http://www.dcc.ufrj.br/~fabiom/comp Análise Sintática A análise sintática agrupa os tokens em uma árvore sintática de acordo com a estrutura do programa
Compiladores - Gramáticas Fabio Mascarenhas - 2013.1 http://www.dcc.ufrj.br/~fabiom/comp Análise Sintática A análise sintática agrupa os tokens em uma árvore sintática de acordo com a estrutura do programa
Análise de Algoritmos
 Análise de Algoritmos Parte 1 Prof. Túlio Toffolo http://www.toffolo.com.br BCC202 Aula 04 Algoritmos e Estruturas de Dados I Qual a diferença entre um algoritmo e um programa? Como escolher o algoritmo
Análise de Algoritmos Parte 1 Prof. Túlio Toffolo http://www.toffolo.com.br BCC202 Aula 04 Algoritmos e Estruturas de Dados I Qual a diferença entre um algoritmo e um programa? Como escolher o algoritmo
MD Lógica de Proposições Quantificadas Cálculo de Predicados 1
 Lógica de Proposições Quantificadas Cálculo de Predicados Antonio Alfredo Ferreira Loureiro loureiro@dcc.ufmg.br http://www.dcc.ufmg.br/~loureiro MD Lógica de Proposições Quantificadas Cálculo de Predicados
Lógica de Proposições Quantificadas Cálculo de Predicados Antonio Alfredo Ferreira Loureiro loureiro@dcc.ufmg.br http://www.dcc.ufmg.br/~loureiro MD Lógica de Proposições Quantificadas Cálculo de Predicados
Capítulo 7. Expressões e Sentenças de Atribuição
 Capítulo 7 Expressões e Sentenças de Atribuição Introdução Expressões são os meios fundamentais de especificar computações em uma linguagem de programação Para entender a avaliação de expressões, é necessário
Capítulo 7 Expressões e Sentenças de Atribuição Introdução Expressões são os meios fundamentais de especificar computações em uma linguagem de programação Para entender a avaliação de expressões, é necessário
MA21 (2015) - Teste - Gabarito comentado. Problema 1 (OBM 2005) Na sequência de números
 - Teste - Gabarito comentado. Problema 1 (OBM 2005) Na sequência de números") MA21 (2015) - Teste - Gabarito comentado Problema 1 (OBM 2005) Na sequência de números 1, a, 2, b, c, d,... dizemos que o primeiro termo é 1, o segundo é a, o terceiro é 2, o quarto é b, o quinto é c e
MA21 (2015) - Teste - Gabarito comentado Problema 1 (OBM 2005) Na sequência de números 1, a, 2, b, c, d,... dizemos que o primeiro termo é 1, o segundo é a, o terceiro é 2, o quarto é b, o quinto é c e
Transformada de Discreta de Co senos DCT
 Transformada de Discreta de Co senos DCT O primeiro passo, na maioria dos sistemas de compressão de imagens e vídeo, é identificar a presença de redundância espacial (semelhança entre um pixel e os pixels
Transformada de Discreta de Co senos DCT O primeiro passo, na maioria dos sistemas de compressão de imagens e vídeo, é identificar a presença de redundância espacial (semelhança entre um pixel e os pixels
3 Redes Neurais Artificiais
 3 Redes Neurais Artificiais 3.1. Introdução A capacidade de implementar computacionalmente versões simplificadas de neurônios biológicos deu origem a uma subespecialidade da inteligência artificial, conhecida
3 Redes Neurais Artificiais 3.1. Introdução A capacidade de implementar computacionalmente versões simplificadas de neurônios biológicos deu origem a uma subespecialidade da inteligência artificial, conhecida
TEOREMA FUNDAMENTAL DA ARITMÉTICA: APLICAÇÕES
 4. TEOREMA FUNDAMENTAL DA ARITMÉTICA: APLICAÇÕES 1). Achando os divisores de um número natural 2). Quantidade de divisores de um número natural 3). Decidindo se um número natural divide outro 4). Extrema
4. TEOREMA FUNDAMENTAL DA ARITMÉTICA: APLICAÇÕES 1). Achando os divisores de um número natural 2). Quantidade de divisores de um número natural 3). Decidindo se um número natural divide outro 4). Extrema
Bioinformática. Conceitos Fundamentais de Biologia Molecular. Paulo Henrique Ribeiro Gabriel phrg@ufu.br
 Bioinformática Conceitos Fundamentais de Biologia Molecular Paulo Henrique Ribeiro Gabriel phrg@ufu.br Faculdade de Computação Universidade Federal de Uberlândia 24 de agosto de 2015 Paulo H. R. Gabriel
Bioinformática Conceitos Fundamentais de Biologia Molecular Paulo Henrique Ribeiro Gabriel phrg@ufu.br Faculdade de Computação Universidade Federal de Uberlândia 24 de agosto de 2015 Paulo H. R. Gabriel
x exp( t 2 )dt f(x) =
dt f(x) =") INTERPOLAÇÃO POLINOMIAL 1 As notas de aula que se seguem são uma compilação dos textos relacionados na bibliografia e não têm a intenção de substituir o livro-texto, nem qualquer outra bibliografia Aproximação
INTERPOLAÇÃO POLINOMIAL 1 As notas de aula que se seguem são uma compilação dos textos relacionados na bibliografia e não têm a intenção de substituir o livro-texto, nem qualquer outra bibliografia Aproximação