UNIVERSIDADE DE BRASÍLIA. Estudo da Ligação de Hidrogênio em Aglomerados de Magnésio
|
|
|
- Manuella Teixeira Furtado
- 5 Há anos
- Visualizações:
Transcrição
1 UNIVERSIDADE DE BRASÍLIA INSTITUTO DE FÍSICA DISSERTAÇÃO DE MESTRADO Estudo da Ligação de Hidrogênio em Aglomerados de Magnésio Por NELSON FERNANDO CHO Brasília-DF, 8 de julho de 2005.
2 DISSERTAÇÃO DE MESTRADO Estudo da Ligação de Hidrogênio em Aglomerados de Magnésio Por Nelson Fernando Cho Orientador Prof. Dr. Paulo Hora Acioli Dissertação submetida ao Instituto de Física da Universidade de Brasília como requisito para a obtenção do grau de Mestre em Física
3 Estudo da Ligação de Hidrogênio em Aglomerados de Magnésio Por Nelson Fernando Cho Dissertação apresentada ao Instituto de Física da Universidade de Brasília como parte dos requisitos necessários para a obtenção do título de Mestre em Física. Aprovada por: Prof. Paulo Hora Acioli (Orientador) IF-UnB Prof. Geraldo Magela e Silva FIS/UnB Prof. João Batista Lopes Martins FIS/UnB 1
4 Brasília, Prof. Sebastião Willian da Silva Coordenador de Pós-Graduação Instituto de Física Universidade de Brasília 2
5 Por mais crítica que seja a situação e as circustâncias em que te encontrares, não te desespere. Nas ocasiões em que tudo inspira temor, nada deves temer. Quando estiveres cercado de todos os perigos, não deves temer nenhum. Quando estiveres sem nenhum recurso, deves contar com todos. Quando fores supreendido, supreende o inimigo. Sun Tzu 3
6 4 A minha família, em especial meu avô.
7 Agradecimentos Meus sinceros agradecimentos ao colega e orientador Prof. Paulo Hora Acioli pela paciência, seriedade, apoio e tantas outras coisas. Aos Professores Ricardo Gargano, Geraldo Magela e Antônio Carlos Pedroza pela disponibilidade de sempre me ajudar. À minha família por tudo o que sou e em especial ao meu avô por ser a pessoa que ele é. Aos professores do Instituto de Física que contribuíram para a minha formação. Aos colegas Matheus, Luiz, Alex, Luana, Marcelo, Fábio, Nanderson, Jonathas, e tantos outros (pessoal do conjunto 1) que tanto me ajudaram durante esse período. À CAPES, pela bolsa de mestrado, que possibilitou este trabalho. E acima de tudo, à Deus. 5
8 Resumo Por se tratar de um elemento abundante e de uma fonte de energia limpa, o hidrogênio se tornou um forte candidato a substituir os combustíveis fósseis em um futuro próximo. Desta forma, o estudo de armazenamento de hidrogênio se torna de suma importância. Os aglomerados de magnésio são bons candidatos para o armazenamento de hidrogênio pelo fato dele ser um metal leve e a grande capacidade de absorção de hidrogênio por superfícies metálicas. Este trabalho tem como finalidade o estudo das ligações do hidrogênio em pequenos aglomerados de magnésio para compreender seu mecanismo de ligação através do uso da Teoria do Funcional da Densidade. A forma preferencial de armazenamento do hidrogênio nos aglomerados de magnésio é a atômica. Determinamos que: existe rearranjo estrutural nos aglomerados menores após o acréscimo de hidrogênio; os aglomerados Mg n H 2n, n 4, saturados assumem a forma linear; os sítios preferenciais para ligação dos hidrogênios são as arestas externas do aglomerado e os átomos de superfície. 6
9 Abstract Due to the fact that hydrogen is an abundant element and a source of clean energy, it has become a solid candidate to replace fossil fuels in a near future. Therefore, the study of hydrogen storage is very important. Magnesium clusters are good candidates to store hydrogen because magnesium is a light metal and the great capacity of hydrogen absorption by metallic surfaces. The purpose of this work is to study the hydrogen bonding with small magnesium clusters through the usage of the Density Functional Theory (DFT). The preferential form of hydrogen bonding in the magnesium clusters is the atomic one. We determined that: there may be structural rearrangement after the addition of hydrogen; the saturated Mg n H 2n, n 4, magnesium clusters have a linear shape; the preferential sites to bind the hydrogen atoms are the edges of the clusters and the surface atoms. 7
10 Sumário 1 Introdução 14 2 Metodologia O Problema Eletrônico Fundamentos da Teoria do Funcional da Densidade Teoremas de Hohemberg-Kohn Equações de Kohn-Sham Aproximação Becke-Perdew Pseudopotenciais Pseudopotencial de Wadt-Hay Resultados Mg n MgH m Mg 2 H m
11 3.4 Mg 3 H m Mg 4 H m Mg 5 H m Mg 6 H m Considerações Finais Conclusão 88 A Estruturas Estáveis e Pontos de Cela para os Aglomerados Mg n H m 90 B Orbitais Moleculares dos Aglomerados Mg n H m 99 9
12 Lista de Figuras 3.1 A linha sólida representa a energia de ligação média do magnésio nos aglomerados Mg n (eixo da esquerda), a linha pontilhada representa o caráter-p dos aglomerados Mg n (eixo da direita) Estruturas estáveis encontradas para o MgH m Distribuição eletrônica no a) MgH; b) MgH Distribuição eletrônica no a) MgH 3 ; b) MgH A linha cheia representa a energia de ligação do hidrogênio no MgH m. A linha pontilhada representa a energia de ligação do hidrogênio molecular Estruturas estáveis encontradas para o Mg 2 H m Distribuição eletrônica no a) Mg 2 ; b) Mg 2 H linear; c) Mg 2 H não-linear Distribuição eletrônica no a) Mg 2 H 2 mais estável; b) Mg 2 H 2 na forma de um paralelogramo.; c) Mg 2 H 2 com dois hidrogênios ligados ao mesmo magnésio
13 3.9 Distribuição eletrônica no a) Mg 2 H 3 mais estável; b) Mg 2 H 3 formado a partir do Mg 2 H 2 linear Distribuição eletrônica no Mg 2 H 4 mais estável A linha cheia representa a energia de ligação do átomo de hidrogênio nos aglomerados Mg 2 H m e a linha pontilhada a energia de ligação do hidrogênio molecular nos aglomerados Mg 2 H m Estruturas estáveis encontradas para o Mg 3 H m Distribuição eletrônica no a) Mg 3 linear; b) Mg 3 H linear Distribuição eletrônica no a) Mg 3 linear ; b) Mg 3 H com o hidrogênio ligado a um dos átomos de magnésio; c) Mg 3 H com o hidrogênio ligado a uma das arestas do triângulo Distribuição eletrônica no a) Mg 3 H 2 triangular com os átomos de hidrogênio ligados ao mesmo magnésio; b) Mg 3 H 2 linear com os átomos de hidrogênio ligados ao mesmo magnésio Energia de ligação do hidrogênio no Mg 3 H m Estrutura energeticamente intermediária entre o Mg 4 mais estável e o linear Distribuição eletrônica do a) Mg 4 mais estável; b) Mg 4 H mais estável Evolução da otimização para o Mg 4 H Rearranjo do Mg 4 H 5 e as perdas de energias em cada processo Estruturas mais estáveis para o Mg 4 H m
14 3.22 Possíveis estruturas para o Mg Estruturas mais estáveis para o Mg 5 H m Distribuição eletrônica no a) Mg 5 mais estável; b) Mg 5 H 2 mais estável a) Estrutura mais estável do Mg 6 ; b) Segundo isômero do Mg Estruturas mais estáveis para o Mg 6 H m A linha sólida representa a energia de ligação do primeiro hidrogênio e a pontilhada representa a energia de ligação do segundo hidrogênio Caráter-p dos aglomerados Mg n H m para m = 1,.., Caráter-p dos aglomerados Mg n H m para m = 7, 9, 13, A.1 Outras estruturas de maior energia para o Mg 2 H m, onde ML representa um mínimo local e PC um ponto de cela A.2 Outras estruturas de maior energia para o Mg 3 H m, onde ML representa um mínimo local e PC um ponto de cela A.3 Outras estruturas de maior energia para o Mg 4 H m, m 4, onde ML representa um mínimo local e PC um ponto de cela A.4 Outras estruturas de maior energia para o Mg 4 H m, m > 4, onde ML representa um mínimo local e PC um ponto de cela A.5 Outras estruturas de maior energia para o Mg 5 H m, m 2, onde ML representa um mínimo local e PC um ponto de cela
15 A.6 Outras estruturas de maior energia para o Mg 5 H m, m > 2, onde ML representa um mínimo local e PC um ponto de cela A.7 Outras estruturas de maior energia para o Mg 6 H m, m 2, onde ML representa um mínimo local e PC um ponto de cela A.8 Outras estruturas de maior energia para o Mg 6 H m, m > 2, onde ML representa um mínimo local e PC um ponto de cela B.1 Orbitais atômicos do magnésio e moleculares do MgH B.2 Orbitais moleculares do MgH B.3 Orbitais moleculares do Mg 2 e Mg 2 H para duas geometrias B.4 Orbitais moleculares do Mg 2 H 2 para duas geometrias B.5 Orbitais moleculares do Mg 2 H 3 para duas geometrias B.6 Orbitais moleculares do Mg 3 e do Mg 3 H B.7 Orbitais moleculares do Mg 3 H B.8 Orbitais moleculares do Mg 3 H B.9 Orbitais moleculares do Mg 3 H B.10 Orbitais moleculares do Mg 3 H B.11 Orbitais moleculares do Mg 4 e do Mg 4 H B.12 Orbitais moleculares do Mg 5 e do Mg 5 H
16 Capítulo 1 Introdução Combustíveis fósseis são uma das principais fontes de energia desde a revolução industrial, no século XVIII. Um exemplo de sua importância foi a crise econômica mundial na década de 70, conhecida como a crise do petróleo, gerada devido ao sucessivo aumento do preço do petróleo imposto pela OPEP. Neste período, o valor do barril de petróleo comprado pelo Brasil aumentou de US$3, 86 para US$21, 50. A contínua utilização desta fonte de energia compromete tanto o meio ambiente, devido à emissão de dióxido de carbono (CO 2 ) e outros poluentes, como suas reservas, que são limitadas e localizadas em certas regiões do planeta. A questão ambiental vem sendo amplamente trabalhada no contexto mundial na forma de tratados internacionais, como o Protocolo de Kioto. A preocupação quanto sua escassez está embasada na crescente demanda 14
17 mundial por energia. Para se ter uma idéia, estima-se que esta demanda dobrará até 2050 devido ao crescimento populacional e a industrialização dos países subdesenvolvidos[1]. Torna-se então natural a procura por fontes de energia alternativas, como biomassa, etanol, biodisel ou energia eólica, que podem substituir os combustíveis fósseis, de tal maneira a atender a demanda mundial e não comprometer o meio ambiente. Dentre as diversas fontes de energia alternativas, destaca-se o hidrogênio devido à sua abundância e a sua concentração de energia, de 120.7kJ/g. A extração de energia a partir de hidrogênio é feita através de sua mistura com oxigênio, resultando em uma mistura extremamente inflamável. O saldo desta reação é uma grande quantidade de energia e água, o que não compromete o meio ambiente. Outra forma de obtenção de energia a partir do hidrogênio é a fusão nuclear, que consiste na fusão de dois átomos de hidrogênio para a formação do hélio. Assim como no primeiro caso, a fusão é benigna uma vez que seu resultado é o hélio, além de energia. As principais dificuldades no uso do hidrogênio como fonte de energia são sua obtenção e seu armazenamento. Mesmo sendo abundante, a forma de ocorrência mais freqüente do hidrogênio é em compostos químicos, como a água. A sua remoção a partir destes compostos químicos é então a chave de seu uso como fonte de energia. No caso da água, a obtenção do hidrogênio pode ser feita de duas maneiras: através da eletrólize da água, um processo que necessita de uma 15
18 grande quantidade de energia, ou através do uso de ácido clorídrico, que resulta na formação de compostos poluentes. Um outro problema, motivação deste trabalho, é a existência de um mecanismo eficaz para seu armazenamento. O Departamento de Energia dos Estados Unidos da América (DOE 1 ) tem como meta o uso de células de combustível que tenham 6.5% de sua massa proveniente de hidrogênio armazenado, o que corresponde a uma densidade de 62.5kg/m 3. Por ser muito leve e ter um ponto de liquefação muito baixo (em torno de 20K), mecanismos simples como o armazenamento na forma de gás ou líquido são muito pouco eficientes e trabalhosos. Por exemplo, a temperatura e pressão ambientes, 1kg de hidrogênio na forma gasosa ocupa 11m 3, o que representa uma densidade de 0.01kg/m 3. Existe hoje uma grande quantidade de materiais estudados com a finalidade de armazenar hidrogênio. Dentre eles, pode-se citar nanotubos de carbono dopados com lítio[2], armações de metal-orgânico (MOF 2 )[3], que são materiais extremamente porosos, ou ligas metálicas, como as ligas de magnésio-níquel[4], devido ao alto grau de reatividade do hidrogênio com metais. Os aglomerados de magnésio surgem como boas possibilidades para o armazenamento de hidrogênio. Por se tratar de um alcalino terroso, o magnésio é altamente reativo com o hidrogênio assim como o lítio utilizado na dopagem 1 do inglês, Department of Energy 2 do inglês, Metal-Organic Framework 16
19 dos nanotubos de carbono. Além disto, o magnésio é um elemento muito leve, o que proporciona uma maior densidade de hidrogênio nos aglomerados Mg n H m resultantes. Estudos anteriores[5] mostram fortes indícios de uma transição dos aglomerados de magnésio de estruturas não-metálicas para estruturas metálicas com o aumento do número de átomos nos aglomerados. Espera-se que os aglomerados na fase metálica sejam mais apropriados para o armazenamento. O presente trabalho tem como finalidade a compreensão da natureza do mecanismo de ligação do hidrogênio em pequenos aglomerados de magnésio, assim como as posições preferenciais nos aglomerados de magnésio para a fixação do hidrogênio e sua forma mais propícia para o armazenamento, atômica ou molecular. Estas investigações serão conduzidas utilizando-se da teoria do funcional da densidade (DFT 3 ) na aproximação de gradiente generalizado BP86[6, 7]. Essa escolha foi feita tendo em consideração os bons resultados obtidos nos estudos dos aglomerados de magnésio [5]. Essa dissertação será dividida em 4 capítulos: no segundo, a metodologia e as ferramentas utilizadas serão introduzidas; no terceiro, os resultados obtidos para os diversos hidretos de magnésio estudados são exibidos e o quarto apresenta as conclusões deste trabalho, além das perspectivas para trabalhos futuros. No apêndice A apresentamos outras estruturas estáveis dos aglomerados Mg n H m. No apêndice B apresentamos os orbitais moleculares dos aglomerados Mg n H m. 3 do inglês, Density Functional Theory 17
20 Capítulo 2 Metodologia Utilizamos a DFT para o cálculo de energia dos aglomerados Mg n H m na correção de gradiente generalizado BP86[6, 7] e o pseudopotencial de Wadt-Hay[8] para o tratamento dos elétrons de valência do magnésio. Esta escolha foi baseada em testes[5] realizados em aglomerados de magnésio com até 5 átomos utilizando-se diversos pseudopotenciais e aproximações para o potencial de troca-e-correlação. A próxima seção tem como objetivo introduzir o problema eletrônico e necessidade de um tratamento acurado da correlação eletrônica. A segunda discorre sobre a DFT e a última seção trata do pseudopotencial utilizado neste trabalho. 2.1 O Problema Eletrônico A importância do cálculo de estrutura eletrônica tem aumentado nos últimos tempos. Estudos de bandas de condução e propriedades de ligações moleculares 18
21 são de extrema importância para a confecção de semicondutores e diversos outros materiais. Como conseqüência disso, inúmeros métodos foram desenvolvidos para o tratamento de sistemas compostos atômicos, tendo eles suas vantagens e desvantagens. O foco de muitos desses métodos é encontrar a solução da equação de Schröndiger estacionária e não-relativística de um sistema composto por núcleos e elétrons: (T e + T n + V ee + V nn + V ne )Ψ(x 1,..., x n ) = EΨ(x 1,..., x n ) (2.1) onde T e é a energia cinética dos elétrons, T n é a energia cinética dos núcleos, V ee é o potencial de interação entre os elétrons, V nn é o potencial de interação entre os núcleos, V en é a interação entre os elétrons e os núcleos, E é a energia do sistema e Ψ(x 1,..., x n ) é a função de onda que representa o sistema. A primeira aproximação importante para tratar (2.1) é utilizar a aproximação de Born-Oppenheimer[9]. Ela considera a grande diferença de massa entre os núcleos e os elétrons para que os últimos se movam num campo de núcleos fixos. O resultado dessa aproximação é a separação do problema inicial em dois: um eletrônico e outro nuclear, onde os núcleos se movem no campo médio formado pelos elétrons. A solução do problema eletrônico fornece informações sobre propriedades como ligações moleculares ou energias de ionização e o nuclear fornece 19
22 informações sobre energias translacionais, rotacionais ou vibracionais de átomos. A função de onda para um sistema de n elétrons deve ser antisimétrica com respeito a troca de duas coordenadas x i e x j de dois elétrons quaisquer, satisfazendo-se o princípio de exclusão de Pauli, para a resolução do problema eletrônico, ou seja, Ψ(x 1,.., x i, x j,..., x n ) = Ψ(x 1,.., x j, x i,..., x n ) (2.2) onde x i são as coordenadas espaciais e de spin do elétron i. Uma maneira de se construir funções de onda antisimétricas é escrevê-las como um determinante de uma matriz que tenham como colunas os n spins orbitais χ j, que são as funções de onda que descrevem o elétron j, e como n linhas as coordenadas x i do elétron i. A troca da coordenada x i de dois elétrons corresponde então a uma troca de duas colunas, o que acarreta na mudança de sinal do determinante. Ao se considerar ainda a normalização da função de onda, tem-se o chamado Determinante de Slater: Ψ(x 1,.., x n ) = 1 N! χ 1 (x 1 )... χ n (x 1 )..... χ 1 (x n )... χ n (x n ) (2.3) A aproximação de Hartree-Fock (HF) pode ser feita ao se procurar os me- 20
23 lhores orbitais que minimizem a energia total do sistema utilizando-se o método variacional. O resultado é um sistema de n-equações integro diferenciais que descrevem cada elétron do sistema e são resolvidas de maneira alto-consistente. h(x 1 )χ a (x 1 ) + [ b a [ b a ] dx 2 χ b (x 2 ) 2 r12 1 χ a (x 1 ) ] dx 2 χ b(x 2 )χ a (x 2 )r12 1 χ b (x 1 ) = ɛ a χ a (x 1 ) (2.4) onde h(x 1 ) = ( A ) Z A r ia (2.5) é a energia cinética e energia potencial de atração ao núcleo do elétron 1 e ɛ a é a energia do spin orbital χ a. O segundo termo de (2.4) é o termo de Coulomb e representa o campo médio sentido pelo elétron 1 no spin orbital χ a. O terceiro termo aparece devido à antisimetria da função de onda e é chamado de termo de troca. Ele representa a correlação entre elétrons de mesmo spin. O resultado dessa aproximação fornece cerca de 99% da energia total eletrônica. A priori, isso representa um ótimo resultado. Todavia, a energia de ligação entre átomos é da ordem de 1% da energia total eletrônica. Isso faz com que a aproximação de Hartree-Fock possa vir a ruim no estudo de ligações entre átomos. A diferença entre a energia total do sistema e a energia calculada no Hartree-Fock 21
24 é chamada energia de correlação E c = E exato E HF (2.6) onde E exato é a energia exata do sistema e E HF é a energia encontrada no Hartree- Fock. Para tratar a correlação, outros métodos foram desenvolvidos como a Configuração de Interação (CI)[9], a Aproximação de Pares de Elétrons Independentes (IEPA)[9], a Aproximação de Aglomerados Acoplados (CCA)[9] e a Teoria de Perturbação[9]. Dentre eles, alguns são variacionais mas não são consistentes com o tamanho do sistema, como o CI e o HF, enquanto outros são consistentes com o tamanho mas não são variacionais, como o CCA e o IEPA. O grande revés destes métodos é o seu alto custo computacional A DFT será empregada neste trabalho por ser um método variacional e consistente com o tamanho do sistema que trata a energia de correlação, além de ser computacionalmente factível. O DFT foi estabelecido como teoria em 1964, quando Hohemberg e Kohn[10] mostraram uma relação unívoca entre a densidade eletrônica e um potencial externo baseado na idéia do modelo de Thomas- Fermi[11, 12], criado na década de vinte. Dessa forma, torna-se viável a utilização da densidade eletrônica no lugar da função de onda no tratamento do sistema. Com isso, pode-se reescrever os operadores como funcionais da densidade e o problema 22
25 de 3N graus de liberdade é simplificado a um de apenas três graus. Em 1965, Kohn e Sham[13] formularam um maneira de reaver o tratamento com funções de onda. Para tal, o sistema físico real é mapeado em um sistema de quase-partículas não interagentes com a mesma densidade do real. Ao fazer tal substituição, chega-se a um sistema de equações similar ao obtido no HF. 2.2 Fundamentos da Teoria do Funcional da Densidade A Teoria do Funcional da Densidade utiliza a densidade eletrônica como variável básica para se resolver a equação de Schröndiger, ao invés de funções de onda. Com isso, a energia cinética e as interações de um sistema de N partículas que interagem entre si são descritas como funcionais da densidade. A primeira tentativa de utilizar esses funcionais originou o modelo de Thomas-Fermi[11, 12], na década de vinte. Entretanto, para se estabelecer a viabilidade do uso da densidade eletrônica na descrição do sistema, é necessária a prova que tal substituição é possível e existam funcionais exatos. Os trabalhos de Hohemberg e Kohn[10] demonstraram tal viabilidade. 23
26 2.2.1 Teoremas de Hohemberg-Kohn Teorema 1. O potencial externo de um sistema é determinado pela densidade eletrônica do sistema, a menos de uma constante. Seja a Hamiltoniana eletrônica dada por: Ĥ = ˆT + ˆV ee + ˆV ext (2.7) onde ˆT é a energia cinética dos elétrons, ˆVee é o potencial de interação entre os elétrons e ˆV ext é um potencial externo. A hamiltoniana descrita em (2.7) mostra que um sistema eletrônico difere de outro devido à forma de ˆV ext e ao número de elétrons. O termo ˆT + ˆV ee = ˆF que representa a energia cinética dos elétrons e as interações entre eles é sempre da mesma forma. Desta forma, pode-se considerar este operador ˆF como sendo universal. Suponha então a existência de dois sistemas, 1 e 2, com um mesmo número de elétrons. Os potenciais externos ˆV 1 e ˆV 2 diferem por mais de uma constante, mas geram a mesma densidade eletrônica ρ(r). Para cada sistema, haverá uma função de onda não-degenerada que descreve seu estado fundamental, Ψ 1 para o primeiro e Ψ 2 para o segundo, que geram a mesma densidade. Utilizando-se Ψ 2 para o cálculo da energia média do sistema 1, E 1, e Ψ 1 para a determinação da 24
27 energia média de 2, E 2, teremos: E 1 = Ψ 2 ˆT + ˆVee + ˆV 1 Ψ2 (2.8) e E 2 = Ψ 1 ˆT + ˆVee + ˆV 2 Ψ1 (2.9) Como as densidades geradas por Ψ 1 e Ψ 2 são iguais e os estados fundamentais dos sistemas são não-degenerados, tem-se das equação (2.7) e do princípio variacional: Ψ1 ˆT + ˆVee Ψ1 + ρ(r)v 1 (r)dr Ψ 2 ˆT + ˆVee Ψ2 + Ψ1 ˆT + ˆVee Ψ1 Ψ2 ˆT + ˆVee Ψ2 ρ(r)v 1 (r)dr (2.10) e Ψ2 ˆT + ˆVee Ψ2 + ρ(r)v 2 (r)dr Ψ 1 ˆT + ˆVee Ψ1 + Ψ2 ˆT + ˆVee Ψ2 Ψ1 ˆT + ˆVee Ψ1 ρ(r)v 2 (r)dr (2.11) onde o lado direito das desigualdades representam a energia do estado fundamental do sistema 1 e 2, respectivamente. As relações (2.10) e (2.11) são satisfeitas se e somente se as funções de onda 25
28 Ψ 1 e Ψ 2 forem iguais, o que significa que os potenciais ˆV 1 e ˆV 2 são iguais a menos de uma constante. Isso contradiz a suposição inicial imposta aos potenciais externos e garante a existência de uma relação unívoca entre o potencial e a densidade ρ(r), sendo o primeiro teorema então provado. Como a densidade eletrônica gerada por esses potenciais também fornece a quantidade de elétrons do sistema, chega-se a conclusão de que ela descreve univocamente um sistema eletrônico com um estado fundamental não-degenerado. Desta forma, podemos escrever a função de onda e a energia do sistema como funcionais da densidade: Ψ = Ψ[ρ] E = Ψ ˆT + ˆV ee + ˆV 2 Ψ = Ψ[ρ] ˆT + ˆVee + ˆV 2 Ψ[ρ] E[ρ] = T [ρ] + V ee [ρ] + V ext [ρ] (2.12) Teorema 2. A energia do estado fundamental pode ser obtida variacionalmente: a densidade que miniminiza a energia total é a densidade do estado fundamental. Do princípio variacional, sabemos que para um sistema com o estado fundamental descrito por Ψ = Ψ[ρ] temos: Ψ ˆT + ˆVee + ˆV ext Ψ Ψ ˆT + ˆVee + ˆV ext Ψ (2.13) 26
29 Como visto anteriormente em (2.12), a função de onda e os operadores em (2.13) podem ser escritos como funcionais da densidade. Desta forma, Ψ = Ψ [ρ ] e a desigualdade acima é reescrita como: T [ρ] + V ee [ρ] + V ext [ρ] T [ρ ] + V ee [ρ ] + V ext [ρ ] E[ρ] E[ρ ] (2.14) A relação (2.14) é o princípio variacional em função da densidade eletrônica do sistema. Ao se impor um número fixo de partículas no sistema, pode-se então minimizar a energia do sistema utilizando-se o método dos multiplicadores de Lagrange: δe[ρ(r)] ρ(r) δe[ρ(r)] µn = 0 δρ(r) µ δ ρ(r)dr = 0 δρ(r) δf [ρ(r)] + V ext (r) = µ (2.15) δρ(r) A principal dificuldade para o uso da densidade como variável básica é encontrar a forma exata para o funcional F [ρ((r))] da energia. O modelo de Thomas-Fermi[11, 12], por exemplo, considera diversas hipóteses para a construção do funcional que descreve a energia cinética: correções relativísticas desprezíveis, elétrons distribuídos uniformemente num espaço de fase de dimensão seis, entre outras. 27
30 2.2.2 Equações de Kohn-Sham Para contornar o problema de se encontrar funcionais para a energia do sistema, Kohn e Sham[13] elaboraram um modelo no qual o sistema eletrônico em questão é substituído por um outro composto por partículas não-interagentes, que possuí a mesma densidade do sistema real. Ao impor densidade idênticas, o primeiro teorema de Hohemberg-Kohn garante que o mesmo estado eletrônico será estudado, sendo a energia cinética totalmente conhecida. O funcional da energia do sistema não-interagente é escrito como em (2.12) para V ee [ρ] = 0: E[ρ(r)] = T s [ρ(r)] + E H [ρ(r)] + E XC [ρ(r)] + υ ext (r)ρ(r)dr E[ρ(r)] = F [ρ(r)] + υ ext (r)ρ(r)dr (2.16) E H [ρ(r)] = 1 2 ρ(r)ρ(r ) dr dr (2.17) r r onde T s [ρ(r)] é o funcional da energia cinética de um elétron livre, E H [ρ(r)] é a energia eletrostática clássica ou termo de Hartree, υ ext é um potencial externo e E XC [ρ(r)] um termo que engloba as diferenças entre o sistema real e o sistema de Kohn-Sham, chamado de energia de troca e correlação. Usando o método dos multiplicadores de Lagrange na equação (2.16) e 28
31 lembrando-se da definição (2.17), tem-se um resultado similar ao obtido em (2.15): δt s [ρ(r)] δρ(r) δf [ρ(r)] + υ ext ρ(r)dr δρ(r) = µ δt s [ρ(r)] + δe H[ρ(r)] + υ ext (r) δρ(r) δρ(r) = µ + υ H (r) + υ XC (r) + υ ext (r) = µ δt s [ρ(r)] δρ(r) + υ KS (r) = µ (2.18) onde υ xc (r) = δe XC[ρ(r)] δρ(r) (2.19) e υ KS (r) = υ H (r) + υ XC (r) + υ ext (r) (2.20) O termo (2.20) é chamado de potencial de Kohn-Sham. Define-se a função de onda Kohn-Sham, φ, como aquela que miniminiza a energia cinética do sistema não-interagente. O artifício de mapear o sistema real em um fictício possibilita o tratamento do problema através da solução de N equações de uma partícula, chamadas de equações de Kohn-Sham: ( 1 ) 2 2 s + υ KS (r) φ i (r) = ɛ i φ i (r) (2.21) 29
32 ρ(r) = N φ i (r) 2 (2.22) i=1 onde ɛ i é a energia associada à função de onda Kohn-Sham φ i (r). A formulação do sistema de equações é exata, gerando a densidade eletrônica do estado fundamental do sistema real. Entretanto, o funcional da energia de trocae-correlação exato não é conhecido. Definindo-se este funcional, é possível resolver o sistema de equações formado pelas N equações de Kohn-Sham de uma partícula. A primeira aproximação para a energia de troca-e-correlação é chamada Aproximação de Densidade Local (LDA 1 )[10, 14, 15] baseada em um sistema conhecido: o gás de elétrons homogêneo. Nela o sistema eletrônico é separado em diversas partes, onde se considera a densidade eletrônica ao redor de um ponto r constante. Assim, a energia de troca-e-correlação é escrita como a soma da energia de troca-e-correlação de diversos sistemas formados por gases de elétrons homogêneos E XC [ρ(r)] = ε XC [ρ(r)]ρ(r)dr. (2.23) O passo seguinte para uma nova energia de troca-e-correlação levou a chamada Aproximação por Expansão de Gradiente (GEA 2 )[10]. Ela consiste na expansão até a primeira ordem da energia de troca-e-correlação. Seus resultados não foram 1 do inglês, Local Density Approximation 2 do inglês, Gradient Expansion Approximation 30
33 satisfatórios, porém serviram como ponto de partida para uma outra aproximação. A Aproximação de Gradiente Generalizado (GGA 3 )[16] melhora a GEA ao se adicionar um parâmetro de corte na GEA para que ela funcione. Estas aproximações são amplamente utilizadas atualmente Aproximação Becke-Perdew 86 Utilizamos a correção de gradiente generalizado Becke-Perdew 86 (BP86) para a energia de troca-e-correlação, onde a energia de troca usada é a desenvolvida por A. D. Becke[6] e a de correlação por John P. Perdew[7]. Esta escolha foi baseado em bons resultados obtidos no cálculo de estrutura eletrônica dos aglomerados de magnésio[5]. A energia de troca obtida por Becke[6] corrige a LDA de tal modo a manter o comportamento assintótico da energia de troca correto, da forma de 1/r. A primeira tentativa, chamada de correção de gradiente de menor ordem (LGC), é determinada exclusivamente por uma análise dimensional: E LGC X = E LDA X β α ( ρα ) 2 dr (2.24) ρ 4/3 α onde o índice α indica o spin do elétron e β é uma constante. Os principais problemas desta correção são a divergência assintótica da energia de troca e a definição do valor da constante β. Para se tentar corrigir estes 3 do inglês, Generalized Gradient Approximation 31
34 problemas, foi introduzido um outro funcional com base semi-empírica[17] de tal modo a se manter a dimensão de energia e obter (2.24) para pequenos gradientes de densidade. Sendo o comportamento assintótico exato dado por: lim r U α X = 1 r (2.25) onde U α X é o potencial de Coulomb do buraco de troca. Baseando-se no decaimento exponencial das funções de onda de Hartree- Fock, pode-se escrever a energia de troca para r como: E X = 1 2 α ρ α U α Xdr (2.26) O comportamento assintótico da densidade também é conhecido[18]: lim ρ α = e aαr (2.27) r onde a constante a α é relacionada ao potencial de ionização do sistema. Becke propôs o seguinte funcional para a energia de troca que reproduz os comportamentos assintóticos descritos em (2.26) e (2.27): E X = E LDA X β α ρ 4/3 α x 2 α dr (2.28) (1 + 6βx α sinh 1 x α ) 32
35 onde β é uma constante e x α = ρ α ρ 4/3 α (2.29) Ao se expandir sinh 1 = x α em Taylor, tem-se a expressão (2.24) para pequenas variações do gradientes de densidade. A constante β é determinada por um ajuste de mínimos quadrados para dados de cálculos Hartee-Fock exatos, sendo seu valor β = A energia de correlação desenvolvida por Perdew[7] tem como base a energia de troca-e-correlação de Langreth e Mehl(LM)[19]. Neste modelo, a energia de troca-e-correlação é escrita como uma expansão em termos de vetores de onda de Fermi k F = (3π 2 ρ) 1/3. Este funcional também considera a redução ao LDA para pequenas variações de gradientes de densidade e tem a seguinte forma EXC LM = EXC LDA + ( ) dr ρ 2 ρ 4/3 ( 2e F 7 ) 9 (2.30) onde F = ρ 2 /ρ 7/6. Ao definir a energia de troca de um modo menos usual[20], Langreth e Mehl encontraram a energia de correlação como sendo a diferença entre (2.30) e a energia de troca por eles separada. Assim como no formalismo de Kohn-Sham, a energia de troca é obtida a partir de um sistema de partículas não-interagentes com a mesma densidade do sistema real, denotada pelo índice L. Desta forma, a 33
36 energia de troca é dada por: E LM X = Ψ L 1 2 e 2 i,j,i j r Ψ L ij E Coul (2.31) onde Ψ L é a função de onda que descreve a densidade do sistema real, não sendo a função de Hartree-Fock necessariamente, e E Coul é a energia Coulombiana E Coul = e2 2 ρ(r)ρ(r ) r r dr dr (2.32) O resultado desta separação se reduz à LDA no limite de pequenos gradientes, mas não a GEA. Além disso, a energia de correlação encontrada não vai além da RP A[21]. Para corrigir tais problemas, considera-se a energia de troca definida no Hartree-Fock para definir a energia de correlação e a suceptibilidade dinâmica de um sistema interagente. A energia de correlação proposta por Perdew é: E C [ρ, ρ ] = ρ(r)ɛ C (ρ, ρ )dr + d 1 e Φ C(ρ) ρ 2 ρ 4/3 dr (2.33) onde ρ(r) = ρ + ρ, ζ = (ρ ρ )/ρ, ρ = (4πr 3 S/3) 1, α = , β = 7.389x10 6, γ = 8.723, δ = e C(ρ) = αr2 S + βr S 1 + γr S + δrs βrs 3 ( ) 5/3 ( ) 5/3 1/2 d = 2 1/3 1 + ζ 1 ζ (2.34) (2.35) 34
37 Φ = 1.745f[C( )/C(ρ)] ρ 7/6 ρ (2.36) O parâmetro de corte f é obtido através da parametrização dos resultados de Ceperley-Adler[15] e seu valor é f = Esta correção tem como principais vantagens a reprodução da LDA para pequenas variações do gradiente da densidade e a preocupação com sistemas pouco densos. 2.3 Pseudopotenciais O cálculo em estrutura eletrônica é de difícil tratamento para um problema de muitos corpos que interagem entre si. A maior contribuição para a energia total de um sistema eletrônico vem dos estados mais próximos ao núcleo. Entretanto, propriedades como a energia de ligação de átomos e a eletroafinidade correspondem a uma pequena fração da energia total do sistema ( ) e são determinadas pelos orbitais mais externos. Uma classificação usual dos elétrons de um átomo é considerar os elétrons mais internos como sendo de caroço e os elétrons mais externos como de valência. Os elétrons de valência são os que participam ativamente das ligações e são fracamente ligados ao núcleo enquanto que os de caroço são fortemente ligados e são pouco afetados pelo meio ao qual o átomo está sujeito. Os elétrons de valência são os principais responsáveis pela periodicidade dos elementos e de propriedades 35
38 como ligações químicas ou potenciais de ionização. Esta divisão faz com que o tratamento dos elétrons de valência possa ser simplificado ao eliminar os elétrons de caroço que são fortemente ligados ao núcleo. Desta maneira, os elétrons de valência estão sujeitos a um potencial efetivo do núcleo atômico mais os elétrons de caroço. Esta simplificação pode ser feita ajustando-se dados experimentais ou através de métodos ab-initio. A separação entre elétrons de caroço e de valência em um cálculo pode ser feita de duas maneiras. Na primeira, chamada de aproximação de caroço congelado, os elétrons de caroço são tratados como estando totalmente estáticos e geram o potencial sentido pelos elétrons de valência. Devido ao princípio de exclusão, é necessária a ortogonalidade entre os orbitais de caroço e os de valência. Esta exigência faz com que os ganhos computacionais não sejam muito grandes uma vez que as integrais dos elementos formados por cruzamentos entre os orbitais de valência e de caroço deverão ser ainda avaliadas. Na segunda maneira, o complexo formada pelos elétrons de caroço e o núcleo atômicos é substituída por um potencial efetivo, chamado de pseudopotencial, que é sentido pelos elétrons de valência. Desta maneira, o ganho computacional é muito maior pois não são avaliadas as integrais que surgem na primeira maneira já mencionada. O princípio de exclusão é inserido neste contexto na forma de um potencial repulsivo que impede que os elétrons de valência colapsem. Assim, os elétrons de valência podem ser tratados explicitamente onde o potencial externo é 36
39 o próprio pseudopotencial. Os Pseudopotenciais de norma conservada garantem que a função de onda φ(r) gerada pelo pseudopotencial tenha a forma exata da função de onda real para uma região fora de uma esfera de raio R, uma vez que a quantidade Q = 1 R φ 2 (r)dr (2.37) φ 2 (R) 0 é mantida constante tanto para a função de onda real quanto para a função de onda gerada pelo pseudopotencial. Existem várias maneiras de se construir pseudopotenciais. Mesmo assim, estes procedimentos devem obedecer os mesmos requerimentos descritos por Hamman, Schlüter e Chiang[22] As energias dos átomos com todos os elétrons e os pseudo-átomos devem ser iguais. Os orbitais normalizados dos átomos com todos os elétrons e pseudo-átomos são iguais a partir de um raio de corte r c. Estas duas propriedades implicam em: A integral de 0 a r c das densidades de carga para ambos os casos devem ser iguais. Os elétrons de valência no pseudo-átomo tem as mesmas propriedades de 37
40 espalhamento do que no átomo com todos os elétrons. Estas condições garantem o que é conhecido como transferabilidade do pseudopotencial, isto é, o pseudopotencial se comporta como o átomo real em ambientes químicos diferentes Pseudopotencial de Wadt-Hay O trabalho realizado por Bachelet et al.[22] gera pseudopotenciais que funcionam para cálculos locais de densidade do H ao Pu. Wadt e Hay[8] utilizam um método no qual o efeito de acoplamento spin-órbita é considerado nos cálculos que consideram todos os elétrons e os elétrons de valência. Deste ponto em diante, um procedimento análogo ao descrito na subseção anterior é empregado para a construção do pseudopotencial de Wadt-Hay. Inicialmente, os orbitais atômicos gerados numericamente são separados em orbitais de valência e de caroço. Funções de onda Hartree-Fock são então geradas para os orbitais de valência, φ l, de momentos angulares l que variam de 0 à L, onde L é tipicamente maior que o mais alto l de qualquer orbital de caroço. Assim, os orbitais de valência que consideram todos os elétrons são transformadas em pseudo-funções de onda suaves φ l que são iguais às verdadeiras funções de onda na região de valência. Da inversão das equações de Schröndiger para uma partícula utilizando-se os pseudo-orbitais φ l, gera-se os potenciais numéricos U l para todos os momentos angulares l. O potencial total é então dado em termos do operador 38
41 de projeção P l = l >< l por L 1 U(r) = U L (r) + [U l U L ]P l (2.38) Por conveniência computacional, obtém-se uma forma analítica para U(r) pelo ajuste das seguintes expressões l=0 r 2 [U l U L ], l = 0, 1,..., L 1 (2.39) r 2 [U L N c /r], l = L (2.40) em gaussianas da forma d k r n k exp( ζ k r 2 ) (2.41) k onde N c é o número de elétrons de valência. Para elementos pesados (Z > 36), o procedimento aqui descrito sofre uma pequena modificação, onde funções de onda relativísticas Hartree-Fock de Cowan e Griffin[23] são empregadas. A escolha dos orbitais de valência dos elementos foi feita, com uma única exceção, considerando-se os orbitais s e p mais externos. Para o magnésio, os parâmetros ζ k e d k são dados para cada n k conforme os valores da tabela
42 n k ζ k d k potencial d potencial s-d potencial p-d Tabela 2.1: Valores dos parâmetros ζ k e d k para um dado n k para o átomo de magnésio 40
43 Capítulo 3 Resultados Os aglomerados Mg n H m foram estudados empregando-se a DFT na aproximação de gradiente generalizado BP 86[6, 7] juntamente com o pseudopotencial de Wadt-Hay[8] e sua base relacionada. O procedimento adotado para a busca de hidretos estáveis consiste no uso de aglomerados Mg n neutros mais estáveis[24] como ponto de partida. Adicionamos a estas estruturas hidrogênios também neutros, tanto na forma atômica quanto na molecular. O composto, cuja sua multiplicidade consideramos 1 para uma quantidade par de átomos de hidrogênio e 2 para uma quantidade ímpar, então relaxa para uma estrutura estável ou para uma estrutura de transição. A forma mais estável encontrada a partir deste procedimento é reutilizada como ponto de partida para a adição de mais um hidrogênio com o objetivo de reduzir o espaço de busca das geometrias dos aglomerados. Repetimos este procedimento até a saturação do aglomerado. O objetivo deste método 41
44 é determinar as posições preferenciais dos átomos de hidrogênio nos aglomerados Mg n e a forma em que ele é armazenado, atômico ou molecular, e o número de hidrogênios que podem ser ligados ao aglomerado inicial. Os cálculos foram realizados em um Pentium 4-2GHz e um Athlon XP GHz. O tempo de cem passos de otimização da geometria dos aglomerados Mg n H m é de cerca de alguns minutos para os menores aglomerados e de horas para aglomerados maiores, dependendo da simetria inicial do aglomerado. Estamos interessados em obter a energia de ligação dos diversos fragmentos que formam os aglomerados. A energia de ligação de um fragmento X em um complexo formado por n fragmentos do tipo X e m fragmentos do tipo Y e a energia de atomização destes sistemas são definidas a partir das seguintes relações: E L (X) = E(X n 1 Y m ) + E(X) E(X n Y m ) (3.1) E A (X n Y m ) = ne(x) + me(y ) E(X n Y m ) (3.2) onde as energias E(X n 1 Y m ), E(X) e E(X n Y m ) são as energias obtidas dos aglomerados mais estáveis encontrados. A partir destas energias podemos entender os processos de ligação, ou de maneira equivalente os processos de fragmentação, dos aglomerados Mg n H m. 42
45 Outra quantidade que pode ajudar a elucidação dos sítios preferenciais de ligação é a contribuição dos orbitais do tipo p na carga total de valência dos aglomerados. Além disso, a natureza dos orbitais de fronteira são cruciais para a ligação dos átomos de hidrogênio aos aglomerados. Para o entendimento correto da ligação dos hidrogênios aos aglomerados puros apresentaremos na próxima seção alguns resultados baseados nos estudos de Acioli e Jellinek[24] para Mg n, n=2,.., Mg n Os aglomerados de magnésio apresentam características que fazem dele um excelente candidato para o armazenamento de hidrogênio, como sua alta reatividade com o hidrogênio e o seu baixo peso atômico. Um outro atributo relevante é o comportamento destes aglomerados em função de seu tamanho. Estudos[5] mostram que, com o aumento dos aglomerados Mg n, a natureza das ligações entre os átomos tende a mudar de um sistema Van der Waals fracamente ligado, para o Mg 2, para sistemas mais fortemente ligados com o aumento do tamanho do aglomerado, como mostra a figura 3.1. Esta figura retrata ainda a evolução do caráter-p dos aglomerados Mg n, obtido a partir da análise populacional dos elétrons de valência nos orbitais atômicos, em função de seu tamanho. Para os aglomerados menores, não há uma grande hi- 43
46 bridização entre os orbitais s e p do magnésio. A estabilidade para os pequenos aglomerados é mantida essencialmente através de interações do tipo Van der Waals. Com o aumento do número de átomos no aglomerado, a importância dos orbitais p do magnésio aumenta indicando uma mudança na natureza das ligações entre os magnésios. 0,8 Caráter-p Energia de Ligação 0,35 0,30 Energia de Ligação (ev) 0,6 0,4 0,2 0,25 0,20 0,15 0,10 caráter-p 0,05 0,0 0, número de magnésios (n) Figura 3.1: A linha sólida representa a energia de ligação média do magnésio nos aglomerados Mg n (eixo da esquerda), a linha pontilhada representa o caráter-p dos aglomerados Mg n (eixo da direita). 44
47 3.2 MgH m Na figura 3.2 estão listadas as estruturas estáveis para o aglomerado MgH m. Esses resultados permitem uma compreensão inicial da ligação entre os átomos de hidrogênio e o magnésio. A adição do hidrogênio ao magnésio na forma molecular não se mostrou propícia para o armazenamento, dado que nos casos onde houve formação do H 2, sua energia de ligação é negativa (MgH 2 ) ou ele é fracamente ligado (MgH 4 e MgH 5 ). Uma análise de Mulliken dos compostos MgH e MgH 2 gerados pela associação do hidrogênio atômico revela um compartilhamento eletrônico entre os átomos de 0.68e e 0.78e, respectivamente, como se pode constatar na figura 3.3. Estes resultados mostram um caráter covalente para a ligação entre o hidrogênio e o magnésio. Uma comparação entre o comprimento de ligação, de 1.741Å, e a freqüência do modo normal vibração, de cm 1, calculados para o MgH mostram uma boa concordância com dados espectroscópicos[25], nos quais o comprimento de ligação e a freqüência de vibração para o MgH são tidos como 1.730Å e cm 1, respectivamente. É importante notar que a energia de atomização do MgH 2 com o hidrogênio molecular é 0.053eV maior que a do MgH 2 com átomos de hidrogênio. Isto implica que a energia do sistema com a molécula de hidrogênio é mais baixa que a energia do aglomerado MgH 2 gerado a partir da adição do hidrogênio atômico. Isto se deve ao fato de a interação entre os átomos de hidrogênio ser muito mais forte que 45
48 a interação entre o hidrogênio e o magnésio. As freqüências dos modos normais de vibração do MgH 2 e do hidrogênio molecular listados na tabela 3.1 mostram que as estruturas interagem pouco entre si. Os modos menores indicam a interação entre a molécula de hidrogênio e o magnésio. A energia de ligação desta molécula de hidrogênio no magnésio é muito fraca e portanto o MgH 2 com o hidrogênio atômico ilustrado em 3.3 é mais estável. MgH 2 H Tabela 3.1: Modos vibracionais do MgH 2 com a molécula de hidrogênio e do H 2 em cm 1 A figura 3.4 mostra uma mudança na natureza das ligações descritas anteriormente devido à adição do terceiro hidrogênio. A mudança da carga compartilhada entre dois dos átomos de hidrogênio do MgH 3 e o magnésio, de 0.78e para 0.43e, e o aumento do comprimento de ligação, de 1.72Å para 1.85Å, aponta para um enfraquecimento da ligação do hidrogênio. Neste ponto, o magnésio se encontra totalmente saturado de átomos de hidrogênio já que ao adicionarmos o quarto hidrogênio existe a formação de H 2 molecular ligado fracamente ao MgH 2 ou ao MgH 3. O quarto hidrogênio acrescido ao MgH 3 já não se liga na forma atômica. 46
49 Ocorre, então a formação da molécula de hidrogênio, sendo ela fracamente ligada ao MgH 2. Uma evidência desta fraca ligação é a distribuição eletrônica do MgH 4, ilustrada na figura 3.4, que apresenta valores quase idênticos aos obtidos para o MgH 2. Uma outra evidência é a comparação entre as freqüências dos modos normais de vibração do MgH 4, MgH 2 e H 2, como mostra a tabela 3.2. As primeiras freqüências são referentes à interação entre a molécula de hidrogênio e o MgH 2. MgH 4 MgH 2 H Tabela 3.2: Modos vibracionais do MgH 4, MgH 2 e H 2 em cm 1 Os orbitais atômicos do magnésio podem fornecer informações adicionais acerca do mecanismo de ligação entre o magnésio e o hidrogênio. A partir da figura B.1, nota-se que os orbitais p do magnésio são os grandes mediadores da ligação do MgH. O orbital ocupado de mais alta energia (HOMO 1 ) deste hidreto é praticamente igual ao orbital p do magnésio orientado na direção de aproximação do hidrogênio. 1 do inglês, Highest Occupied Molecular Orbital 47
= 0.044 ev Simetria: C 1 Distância(Mg-H2)=2.582 Å EA=4.888 ev EL(H2)=-0.0002 ev Simetria:C v Distância(Mg-H2): 7.294 Å EL(H)=4.341 ev EA=14.637 ev EL(H2)=0.")
50 EL(H)=1.651 ev EA=1.651 ev Simetria:C v Ligação: Å EL(H)=4.383 ev EA=9.746 ev EL(H2)=0.024 ev Simetria: C 2V Distância(Mg-H2)= Å EL(H)=3.184 ev EA=4.835 ev Simetria:C v EL(H)=0.549 ev EA= ev EL(H2)= ev Simetria: C 1 Distância(Mg-H2)=2.582 Å EA=4.888 ev EL(H2)= ev Simetria:C v Distância(Mg-H2): Å EL(H)=4.341 ev EA= ev EL(H2)=0.002 ev Simetria: C 1 Distância(H2)=5.114 Å EL(H)=0.529 ev EA= ev Simetria: C 2V Figura 3.2: Estruturas estáveis encontradas para o MgH m. Ainda comparando-se os orbitais listados em B.2, percebe-se que agora o orbital HOMO do MgH é o responsável pela ligação do segundo hidrogênio. Novamente, este orbital encontrava-se orientado em uma direção específica e quase não foi alterado devido a presença do hidrogênio. Estes resultados sugerem que a ligação dos hidrogênios é feita em regiões onde existam orbitais orientados que sejam propícios a formar a ligação com os átomos de hidrogênio. As energias de ligação do hidrogênio são apresentadas na figura 3.5. A ligação do hidrogênio é muito forte para um aglomerado com uma quantidade par 48
51 Figura 3.3: Distribuição eletrônica no a) MgH; b) MgH 2. Figura 3.4: Distribuição eletrônica no a) MgH 3 ; b) MgH 4. de átomos de hidrogênio devido às interações entre pares. Observa-se que o orbital HOMO do MgH 2 é muito semelhante a um orbital não ligante da molécula de hidrogênio. Para o MgH 3 e o MgH 5, a quebra da ligação covalente de um dos átomos de hidrogênio é a causa da queda da energia de ligação do hidrogênio. A ligação das moléculas de hidrogênio geradas a partir da saturação do magnésio é muito fraca. Considerando-se apenas as cargas induzidas nos compostos que formam o MgH 2 e MgH 3, chega-se a um valor estimado para a energia de ligação da molécula de hidrogênio de 0.014eV para o MgH 2 e de 0.022eV para o MgH 3. Estes valores correspondem a metade dos valores calculados que são de 0.028eV 49
52 5,0 4,5 4,0 Energia de Ligação (ev) 3,5 3,0 2,5 2,0 1,5 1,0 0,5 0, m (número de hidrogênio) Figura 3.5: A linha cheia representa a energia de ligação do hidrogênio no MgH m. A linha pontilhada representa a energia de ligação do hidrogênio molecular. para o MgH 2 e de 0.044eV para o MgH 3. Nesta seção mostramos que as ligações fortes entre os átomos de hidrogênio e magnésio são do tipo covalente. O sucessivo acréscimo de hidrogênio resultou em uma saturação para o MgH 3. No caso do MgH 4 e MgH 5, há a formação de hidrogênio molecular fracamente ligado ao MgH m 2. Sendo assim, chega-se a conclusão que a forma preferencial de armazenamento do hidrogênio é a atômica. Nas próximas seções tentaremos verificar se esta tendência é mantida para os aglomerados Mg n, para n = 2,.., 6. 50
=2.046 ev EA=2.137 ev EL(Mg)=0.")
=1.723 ev EL(MgH)=0.622 ev EL(H2)=0.032 ev Simetria: C 1 EL(H)=2.563 ev EA=7.701 ev EL(Mg)=2.337 ev EL(MgH)=1.215 ev Simetria: C 2V Figura 3.")
53 3.3 Mg 2 H m As estruturas mais estáveis para o aglomerado Mg 2 H m estão representadas na figura 3.6. Assim como no caso do átomo de magnésio, o hidrogênio molecular não se liga ao dímero Mg 2. Os hidrogênios que se ligam ao Mg 2 para formar o Mg 2 H m se ligam na forma atômica. Outras configurações de maior energia estão listadas na figura A.1. EL(H)=2.046 ev EA=2.137 ev EL(Mg)=0.486 ev EL(MgH)=0.486 ev Simetria: C V EL(H)=3.175 ev EA= ev EL(Mg)=1.129 ev EL(MgH)=3.861 ev Simetria: D 2H EL(H)=3.005 ev EA=5.138 ev EL(Mg)=0.303 ev EL(MgH)=1.834 ev Simetria: D H EL(H)=1.144 ev EA= ev EL(Mg)=1.723 ev EL(MgH)=0.622 ev EL(H2)=0.032 ev Simetria: C 1 EL(H)=2.563 ev EA=7.701 ev EL(Mg)=2.337 ev EL(MgH)=1.215 ev Simetria: C 2V Figura 3.6: Estruturas estáveis encontradas para o Mg 2 H m. Para a melhor compreensão do mecanismo de ligação dos átomos de hidrogênio ao Mg 2 vamos analisar os orbitais de fronteira do dímero e do Mg 2 H. Os orbitais 51
54 indicados na figura B.3 mostram que, assim como no MgH m, o orbital desocupado de menor energia (LUMO 2 ) do dímero é o responsável pela ligação do hidrogênio no Mg 2 H linear. O HOMO do Mg 2 H linear formado pela adição do hidrogênio é praticamente igual ao LUMO do dímero. O mesmo acontece para o ponto de cela Mg 2 H não linear. A diferença é que neste caso o orbital mediador da ligação é o LUMO+1 do dímero. A diferença de estabilidade entre as duas estruturas Mg 2 H se deve aos orbitais mediadores da ligação do hidrogênio no Mg 2. Os orbitais orientados no sentido da ligação, chamados de orbitais σ, formam a ligação simples que é a mais forte das ligações. Já os orbitais perpendiculares, chamados π, mediam as ligações π que são mais fracas. No caso do Mg 2 H linear, a ligação do hidrogênio é mediada pelo orbital σ do dímero, enquanto que a ligação do caso não linear é mediada pelo orbital π. A análise populacional, ilustrada na figura 3.7, revela que a natureza da ligação do hidrogênio no Mg 2 linear é covalente e possuí um comprimento de ligação muito próximo ao obtido para o MgH. A comparação entre estas distribuições de carga mostra que a adição do hidrogênio influi na ligação entre os átomos de magnésio, o que pode ser constatado a partir da variação do seu comprimento de ligação, de 3.6Å para 3.06Å, e de sua energia de ligação, de 0.003eV para 0.486eV. O acréscimo do hidrogênio gera também um pequeno compartilhamento eletrônico, 2 do inglês, Lowest Unoccupied Molecular Orbital 52
55 de 0.14e, entre os átomos de magnésio, fortalecendo então esta ligação. Ao se considerar a distribuição eletrônica do Mg 2 H não linear ilustrada na figura 3.7, nota-se uma diminuição da carga compartilhada em relação ao MgH para 0.22e e um aumento do comprimento de ligação para 2.01Å entre o hidrogênio e um átomo de magnésio. Mesmo assim, podemos considerar essa interação como sendo covalente. Figura 3.7: Distribuição eletrônica no a) Mg 2 ; b) Mg 2 H linear; c) Mg 2 H não-linear. A estrutura do Mg 2 H 2 mais estável ainda é a linear. Os orbitais listados na figura B.3 mostram que o HOMO do Mg 2 H é responsável pela ligação, gerando o HOMO do Mg 2 H 2 linear. A análise populacional deste aglomerado, apresentado na figura 3.8, mostra que a presença dos átomos de hidrogênio fortalecem ainda 53
56 Figura 3.8: Distribuição eletrônica no a) Mg 2 H 2 mais estável; b) Mg 2 H 2 na forma de um paralelogramo.; c) Mg 2 H 2 com dois hidrogênios ligados ao mesmo magnésio. mais a ligação entre os magnésios, diminuindo seu comprimento de ligação para 2.87Å e aumentando o compartilhamento de elétrons para 0.88e. Outras estruturas estáveis para o Mg 2 H 2 são ilustrados na figura A.1. A ligação do segundo hidrogênio no magnésio já ocupado gera a estrutura Mg 2 H 2 ilustrada em A.1 cuja diferença de energia para a estrutura mais estável é 0.241eV. A comparação entre seus modos normais de vibração do Mg 2 H 2 e do MgH 2, listados na tabela 3.3, mostra que tal distinção é possível. Outra evidência é a comparação das distribuições de carga desta geometria e do MgH 2, ilustradas 54
57 Figura 3.9: Distribuição eletrônica no a) Mg 2 H 3 mais estável; b) Mg 2 H 3 formado a partir do Mg 2 H 2 linear. Figura 3.10: Distribuição eletrônica no Mg 2 H 4 mais estável. respectivamente nas figuras 3.8 e 3.2, que são praticamente iguais, e a energia de ligação no magnésio de 0.061eV. A adição de hidrogênio na aresta formada pelos átomos de magnésio passa a ser viável quando ela ocorre em pares, gerando o Mg 2 H 2 listado em A.1 com uma diferença de energia de 0.441eV em relação a mais estável. As ligações dos átomos de hidrogênio à aresta formada pelo dímero são mediadas pelo LUMO+1 55
58 Mg 2 H 2 MgH Tabela 3.3: Modos vibracionais do Mg 2 H 2 e do MgH 2 em cm 1 do dímero. A interação entre os átomos de magnésio é muito pequena, com um compartilhamento de -0.06e, como mostra a figura 3.8. Entretanto, os orbitais moleculares ilustrado na figura B.4 desta estrutura mostra que a interação entre os átomos de hidrogênio e os de magnésio ajuda na estabilização do Mg 2 H 2. O Mg 2 H 3 mais estável não é formado a partir do Mg 2 H 2 linear, mas sim da associação de um par de hidrogênios na aresta formada pelo magnésio. A diferença de energia entre essas estruturas, de 0.620eV, se deve ao fato de a ligação do terceiro hidrogênio ser muito fraca por ser mediada pelos orbitais π do Mg 2 H 2. Ao se alocar dois átomos de hidrogênio na aresta formada pelo magnésio, a interação entre estes átomos faz com que a ligação destes átomos seja mais forte. Os orbitais desta estrutura, apresentados em B.5, mostram que os orbitais HOMO e HOMO-1 apresentam uma grande contribuição da interação entre os hidrogênios em suas formações. A figura 3.9 mostra as distribuições de carga de ambos os casos do Mg 2 H 3. 56
59 Para o Mg 2 H 3 mais estável, a ligação entre os átomos de magnésio é mediada pelos átomos de hidrogênio. No caso do Mg 2 H 3 gerado a partir do Mg 2 H 2 linear, a ligação entre os átomos de magnésio é modificada e o compartilhamento eletrônico em relação ao Mg 2 H 2 linear caí, de 0.88e para 0.57e. Dos orbitais dos Mg 2 H 3 listados em B.5, observa-se que o sítio preferencial é o átomo desocupado no caso do Mg 2 H 3 mais estável, ou o eixo de simetria C 2V para o Mg 2 H 3 gerado a partir do Mg 2 H 2 linear. Em ambos os casos, os orbitais HOMO do Mg 2 H 3 apresentam uma orientação propícia à formação da ligação do quarto hidrogênio. A segunda geometria possível para o Mg 2 H 4 mostra que a interação entre pares de hidrogênio é importante para a estabilidade da estrutura, uma vez que a adição de um terceiro hidrogênio na aresta resultou em uma estrutura com uma energia 0.98eV maior que o caso mais estável. A distribuição eletrônica do isômero mais estável do Mg 2 H 4, ilustrada na figura 3.10 mostra que a ligação entre os átomos de magnésio é mediada pelos átomos de hidrogênio ligados às arestas do Mg 2 e pelo compartilhamento de cargas gerado pelos hidrogênios localizados no eixo principal de simetria. A carga compartilhada diminui de 0.88e para 0.28e ao se comparar o Mg 2 H 4 com o Mg 2 H 2 linear. Esta estrutura está completamente saturada, uma vez que a adição do próximo hidrogênio resulta em hidrogênio na forma molecular e esta é fracamente ligada ao Mg 2 H 3 resultante, assim como no MgH 4 e MgH 5. A comparação dos 57
60 modos vibracionais listados em 3.4 mostra, que de fato, a estrutura pode ser vista como a associação de uma molécula de hidrogênio ao Mg 2 H 3. Mg 2 H 5 Mg 2 H 3 H Tabela 3.4: Modos vibracionais do Mg 2 H 5, Mg 2 H 3 e H 2 em cm 1 A figura 3.11 mostra as energias de ligação do hidrogênio. Como no MgH m, as ligações do hidrogênio são maiores para uma quantidade par de átomos de hidrogênio. A ligação do magnésio aumenta devido à presença do hidrogênio. No Mg 2 H e Mg 2 H 2, o hidrogênio aumenta o compartilhamento de elétrons entre os átomos de magnésio. Para o Mg 2 H 3 mais estável, a mediação da ligação entre os magnésios por parte dos hidrogênios é a responsável pela estabilização. Nos casos do Mg 2 H 3 e do Mg 2 H 4 gerado a partir do Mg 2 H 2 linear, há uma mistura das contribuições da mediação da ligação pelo hidrogênio e o compartilhamento eletrônico 58
61 entre os magnésios. A baixas energias de ligação das moléculas de hidrogênio indicam que também neste caso o hidrogênio prefere se ligar na forma atômica. 3,5 3,0 Energia de Ligação (ev) 2,5 2,0 1,5 1,0 0,5 0, número de hidrogênios (m) Figura 3.11: A linha cheia representa a energia de ligação do átomo de hidrogênio nos aglomerados Mg 2 H m e a linha pontilhada a energia de ligação do hidrogênio molecular nos aglomerados Mg 2 H m. 59
62 3.4 Mg 3 H m As estruturas mais estáveis do Mg 3 H m são apresentadas na figura O interessante é que estas estruturas não são formadas pelo aglomerado Mg 3 mais estável, que corresponde a um triângulo equilátero, mas sim do Mg 3 linear. A diferença de energia entre estas duas geometrias é de 0.193eV. A figura A.2 mostra que a adição do primeiro e do segundo hidrogênio no isômero triangular resulta em aglomerados Mg 3 H e Mg 3 H 2 que estão 0.042eV e 0.010eV acima das estruturas mais estáveis, respectivamente. A baixa energia de ligação do magnésio ao Mg 3 H linear, de 0.356eV, indica que a forma mais provável de obtenção destas estruturas lineares seja a partir da adição do magnésio ao Mg 2 H ao invés de um rearranjo da estrutura triangular devido à adição do hidrogênio, que parece ser mais complicada de ocorrer. Já as estruturas oriundas do Mg 3 triangular são formadas pelo simples acréscimo de hidrogênio ao aglomerado inicial. Podemos compreender o mecanismo de ligação do primeiro hidrogênio aos isômeros do Mg 3 através de seus orbitais de fronteira listados na figura B.6. O LUMO do Mg 3 linear é o responsável pela ligação do hidrogênio assim como no Mg 2 H. No caso do isômero triangular, O LUMO+2 é o mediador da ligação do hidrogênio tanto à aresta quanto ao vértice do triângulo. A comparação entre a distribuição eletrônica nos aglomerados puros e no 60
63 EL(H)=2.265 ev EA=2.423 ev EL(Mg)=0.356 ev EL(MgH)=0.751 ev Simetria: C V EL(H)=2.639 ev EA= ev EL(Mg)=1.862 ev EL(MgH)=1.354 ev Simetria: C 2V EL(H)=2.925 ev EA=5.419 ev EL(Mg)=0.281 ev EL(MgH)=1.630 ev Simetria: D H EL(H)=3.185 ev EA= ev Simetria: C 2V EL(H)=2.807 ev EA=8.225 ev EL(Mg)= ev EL(MgH)=1.436 ev Simetria: C 2V EL(H)=1.724 ev EA= ev EL(H 2)=0.022 ev Simetria: C S EL(H)=3.017 ev EA= ev EL(Mg)= ev EL(MgH)=1.890 ev Simetria: C2v Figura 3.12: Estruturas estáveis encontradas para o Mg 3 H m. Mg 3 H mostra que a adição do primeiro hidrogênio modifica as ligações entre os átomos de magnésio. No caso linear, a figura 3.13 mostra que o acréscimo do hidrogênio induz um compartilhamento de 0.43e entre os átomos de magnésio mais próximos e 0.20e entre os mais afastados. O comprimento de ligação entre esses pares de magnésio também diminui para 3.44Å e 3.15Å, respectivamente. No triângulo, a figura 3.14 mostra que também existe uma indução de 61
64 Figura 3.13: Distribuição eletrônica no a) Mg 3 linear; b) Mg 3 H linear. compartilhamento eletrônico entre os átomos de magnésio. Quando o hidrogênio se liga ao vértice, a ligação entre os átomos de magnésio opostos aumenta para 4.70Å e o compartilhamento eletrônico entre o vértice e os seus vizinhos para 0.04e. No caso do hidrogênio na aresta, a ligação entre os magnésios que formam a aresta passa a ser mediada pelo hidrogênio e a distância entre estes átomos aumenta para 3.72Å. Os possíveis sítios preferenciais para a adição do segundo hidrogênio podem ser obtidos a partir dos orbitais listados nas figuras B.6 e B.7. O HOMO do Mg 3 H sugere que a posição preferencial no caso linear é a extremidade desocupada da linha. De fato, os resultados encontrados mostram que essa é a configuração de energia mais baixa para o Mg 3 H 2. Os outros isômeros lineares listados em A.2 são análogos aos encontrados para o Mg 2 H m. Novamente, os orbitais LUMO+1 e LUMO+2, que geram ligações mais fracas, são os mediadores destas ligações. 62
65 Figura 3.14: Distribuição eletrônica no a) Mg 3 linear ; b) Mg 3 H com o hidrogênio ligado a um dos átomos de magnésio; c) Mg 3 H com o hidrogênio ligado a uma das arestas do triângulo Interessante notar a partir do LUMO e o HOMO-1 do isômero onde o par de átomos de hidrogênio se ligou à mesma aresta, listados na figura B.7, que a interação entre os pares continua presente, assim como no isômero do Mg 2 H 2 com forma de um paralelogramo. Para o Mg 3 H 2 triangular, tanto o HOMO do isômero triangular mais estável quanto o LUMO+1 do segundo mais estável indicam que o Mg 3 H 2 triangular com um hidrogênio ligado a um magnésio e o outro ligado a sua aresta oposta é um 63
66 Figura 3.15: Distribuição eletrônica no a) Mg 3 H 2 triangular com os átomos de hidrogênio ligados ao mesmo magnésio; b) Mg 3 H 2 linear com os átomos de hidrogênio ligados ao mesmo magnésio. forte candidato à estrutura mais estável. De fato, a figura A.2 mostra que este isômero difere 0.010eV do mais estável. A comparação entre as distribuições eletrônicas do isômero triangular com dois hidrogênios no mesmo magnésio, ilustrado em 3.15, e o Mg 3 puro mostra que o comprimento de ligação entre o magnésio ligado aos hidrogênios e seus vizinhos aumenta de 3.28Å para 3.30Å. Isto levanta a suspeita de que a quebra desta estrutura Mg 3 H 2 possa ser possível. A figura 3.15 mostra que as ligações entre os átomos de magnésio no isômero linear com dois hidrogênios ligados à um magnésio não mudaram muito em relação ao aglomerado linear puro, ilustradas na figura Por não haver uma modificação drástica nas ligações entre os átomos de magnésio, suspeita-se que a quebra desta estrutura em um dímero e um MgH 2 seja menos provável que a remoção do átomo de magnésio mais distante, dado que a distância de ligação deste magnésio 64
67 variou pouco (de 3.44Åpara 3.39Å). A tabela 3.5 mostra os modos normais de vibração de ambos os Mg 3 H 2, além do MgH 2 e do dímero Mg 2. No caso do Mg 3 H 2 triangular, a diferença de seu modo vibracional cm 1 em relação ao modo cm 1 do MgH 2 mostra que a quebra pode não ser tão espontânea quanto imaginado. Este modo corresponde à vibração dos átomos de hidrogênio mantendo-se a distância ao magnésio fixa. Mg 3 H 2 triangular Mg 3 H 2 linear MgH 2 Mg Tabela 3.5: Modos vibracionais dos Mg 3 H 2 com dois hidrogênios ligados em um magnésio (triangular e linear), do MgH 2 e do dímero Mg 2 em cm 1 65
68 A comparação entre os modos vibracionais do dímero, de 90.42cm 1, e as do Mg 3 H 2 linear, de 73.61cm 1, mostra uma grande discordância. Este fato indica que a dissociação do dímero pode também não ser trivial. Ao se comparar os modos normais de vibração do Mg 3 H 2 linear e do Mg 2 H 2, apresentados na tabela 3.6, nota-se uma concordância muito maior para este caso. Isto indica que a dissociação do átomo de magnésio seja mais propícia de ocorrer. Mg 3 H 2 Mg 2 H Tabela 3.6: Modos vibracionais do Mg 3 H 2 linear e do Mg 2 H 2 em cm 1 Os orbitais dos aglomerados Mg 3 H 2 listados na figura B.7 servem novamente como ponto de partida para determinar as localizações preferenciais do terceiro hidrogênio. Para o caso linear, a situação é similar a apresentada para o acréscimo do terceiro hidrogênio no Mg 2 H 2. Como mostra a figura 3.12, a estrutura linear mais estável apresenta uma de suas extremidades sem um átomo de hidrogênio. As interações entre os átomos de hidrogênio na aresta, evidenciadas nos orbitais ocupados e no LUMO do Mg 3 H 2 linear mais estável estão ilustradas na figura B.8. 66
69 Outra possível estrutura linear pode ser feita a partir do uso do LUMO do Mg 3 H 2 para a mediação da ligação do hidrogênio. Uma outra forte razão para acreditar na influência da interação entre pares de hidrogênio na estabilidade das estruturas lineares é a comparação da diferença de energia entre as estruturas Mg 3 H 3 lineares listadas em A.2. A estrutura com a outra aresta ocupada difere de 0.166eV da mais estável enquanto que a estrutura com apenas um hidrogênio difere 0.653eV. A comparação entre os modos normais de vibração das estrutura linear mais estável e do Mg 2 H 3, apresentada na tabela 3.7, mostra que os valores são muito parecidos. Este fato indica que a remoção do magnésio isolado seja possível. Mg 3 H 3 Mg 2 H Tabela 3.7: Modos vibracionais do Mg 3 H 3 linear mais estável e do MgH 3 em cm 1 A partir dos orbitais do Mg 3 H 2 triangular mais estável, listados na figura 67
70 B.7, pode-se estimar as posições preferências de ligação do terceiro hidrogênio. O LUMO viabiliza tanto a ligação do hidrogênio em um dos átomos de magnésio disponíveis quanto em uma das arestas disponíveis, que geram estruturas que diferem 0.003eV entre si. De fato, a comparação dos orbitais do Mg 3 H 2 triangular mais estável com os orbitais apresentados na figura B.8 mostra que o LUMO do Mg 3 H 2 é similar ao LUMO dos Mg 3 H 3 triangulares. O HOMO do Mg 3 H 3 com o hidrogênio ligado em um magnésio é parecido com HOMO-1 do Mg 3 H 2. No caso do Mg 3 H 3 com o hidrogênio ligado na aresta, seu HOMO é gerado a partir do HOMO do Mg 3 H 2. A adição do terceiro hidrogênio na face formada pelos átomos de magnésio é mediada pelos LUMO+1 do Mg 3 H 2, o que resulta em interações fracas. A associação do terceiro hidrogênio na aresta já ocupada resulta em um ponto de cela, o que sugere que as arestas do triângulo suportem apenas um átomo. Estas estruturas provavelmente são difíceis de se dissociar em estruturas menores devido ao fato de os hidrogênios mediarem as ligações entre os átomos de magnésio. O mesmo mecanismo de ligação do hidrogênio descreve os demais aglomerados Mg 3 H m gerados a partir da associação do quarto, quinto e sexto hidrogênio. O aglomerado Mg 3 H 4 listado na figura A.2 que difere 0.233eV do mais estável mostrase um bom candidato para uma remoção do magnésio isolado. A comparação dos espectros vibracionais, listados na tabela 3.8, mostra que a maioria dos modos são muito parecidos, estando as grandes diferenças nos modos cm 1, cm 1 68
71 do Mg 2 H 4. Estes modos correspondem a vibrações dos átomos de hidrogênio. Mg 3 H 4 Mg 2 H Tabela 3.8: Modos vibracionais do Mg 3 H 4 linear mais estável e do Mg 2 H 4 em cm 1 Um fato interessante é a formação do aglomerado Mg 3 H 5 que difere 0.321eV mostra que a ligação na face foi possível com a presença de outro hidrogênio na face oposta. Os orbitais do Mg 3 H 5 com hidrogênios na face listados na figura B.10 mostram que o LUMO+2 e o HOMO-1 indicam a interação entre os hidrogênios. Assim como nos aglomerados Mg 2 H m, o Mg 3 H m linear apresenta um ponto onde os sítios preferenciais são todos preenchidos. A partir deste ponto, a adição de mais um átomo de hidrogênio promove a formação de moléculas de hidrogênio fracamente ligados ao Mg 3 H m. A figura 3.16 mostra a energia de ligação do hidrogênio nos aglomerados 69
72 Mg 3 H m. Como já discutido, suspeita-se que a interação entre pares de hidrogênio proporciona uma grande contribuição para a estabilidade dos aglomerados Mg 3 H m. 3,5 Energia de Ligação (ev) 3,0 2,5 2,0 1, número de hidrogênios (m) Figura 3.16: Energia de ligação do hidrogênio no Mg 3 H m. 3.5 Mg 4 H m Os aglomerados Mg 4 H m apresentam resultados muito interessantes. Diferentemente do Mg 3 H m, as estruturas mais estáveis com pequena concentração de hidrogênio não são as lineares. Entretanto, a sucessiva adição de hidrogênio aos aglomerados mostra-se capaz de provocar um rearranjo da estrutura até chegar a um ponto onde as estruturas lineares sejam as mais favoráveis de um ponto de vista energético. 70
73 A energia de ligação média entre os átomos do Mg 4 mais estável é de 0.313eV, valor este muito maior que a energia de ligação média do Mg 3, de 0.140eV, ou do Mg 2, de 0.046eV. Sendo ainda a diferença de energia entre o Mg 4 mais estável e o linear de 0.856eV, podemos esperar que o acréscimo do primeiro hidrogênio não seja capaz de reestruturar o aglomerado de magnésio. Existe ainda uma outra possível geometria para o Mg 4 que energeticamente se encontra entre a estrutura mais estável e a linear, com uma diferença de 0.640eV em relação ao mais estável. Esta estrutura consiste em um triângulo com um magnésio ligado ao seu vértice, como mostra a figura Figura 3.17: Estrutura energeticamente intermediária entre o Mg 4 mais estável e o linear. A formação do Mg 4 H linear é mais fácil de ocorrer a partir da adição de um átomo de magnésio ao Mg 3 H linear, onde a energia necessária para esse processo é de 0.315eV. Outra possibilidade é a obtenção do Mg 4 H linear a partir da adição do dímero Mg 2 ao MgH, onde a energia necessária é de 0.580eV. 71
74 Novamente, os orbitais do Mg 4 mais estável listados na figura B.11 indicam os sítios preferenciais para a adição do hidrogênio. O LUMO+1 mostra que tanto a face do Mg 4, sua aresta e seu vértice são possíveis posições para mediar a ligação com o hidrogênio. Ao se adicionar o primeiro hidrogênio nas três estruturas, observa-se que o Mg 4 H mais estável é o proveniente do tetraedro, como mostra a figura A comparação desta estrutura com o Mg 4 H linear e triangular mostra uma grande diminuição da diferença energética entre essas estruturas. A diferença cai para 0.387eV no caso do Mg 4 H mais estável proveniente da estrutura ilustrada na figura Para a estrutura linear, ela cai para 0.387eV. Esta diminuição mostra que, com a adição do hidrogênio, estas estruturas começam a ser menos desfavoráveis, quando comparadas às diferenças observadas para os aglomerados puros. A figura 3.18 mostra a distribuição eletrônica no Mg 4 puro e no Mg 4 H mais estável, respectivamente. Assim como no Mg 3 H, o acréscimo do hidrogênio modifica as ligações entre os átomos de magnésio, promovendo um aumento do compartilhamento eletrônico entre as arestas mais próximas do hidrogênio de 0.09e para 0.07eV e uma diminuição de 0.06Å em seu comprimento de ligação. A ligação entre os átomos de magnésio passa a ser mediada pelo átomo de hidrogênio que foi adicionado à aresta, conforme verificado para o Mg 3 H m. O acréscimo do segundo hidrogênio mostra que a estrutura mais estável continua sendo a baseada no tetraedro, embora a diferença entre esta e a linear 72
75 Figura 3.18: Distribuição eletrônica do a) Mg 4 mais estável; b) Mg 4 H mais estável. diminua para 0.326eV. A figura A.3 mostra um fato interessante: a estrutura que difere 0.261eV da mais estável apresenta uma forte deformação em relação ao Mg 4 mais estável. A interação entre os átomos de hidrogênio é apontada como principal causa de tal distorção. A estrutura inicial do Mg 4 é rearranjada ao se adicionar um hidrogênio na aresta mais próxima aos hidrogênios já ligados ao Mg 4. Novamente, atribui-se às interação entre os átomos de hidrogênio o motivo de tal rearranjo, como ilustra a figura Como pode se observar, a nova estrutura corresponde ao Mg 4 indicado na figura A partir da adição do quarto hidrogênio, observa-se que as estruturas espaciais encontradas passam a ser energeticamente mais altas. A figura A.3 mostra que as estruturas espaciais encontradas para o Mg 4 H 4 diferem do caso mais estável de 0.477eV. As outras configurações mostram um comportamento muito parecido 73
76 Figura 3.19: Evolução da otimização para o Mg 4 H 3. com o observado para o Mg 3 H m triangular, o que indica o sucesso do mecanismo utilizado para a descrição dos possíveis sítios preferenciais do hidrogênio. Um novo rearranjo acontece no Mg 4 H 5 ao se acrescentar o hidrogênio na aresta da base do triângulo. O resultado é o ponto de cela ilustrado na figura A.4 com uma diferença de 0.564eV em relação ao Mg 4 H 5 mais estável. Esta estrutura relaxa até outro ponto de cela que difere 0.014eV do aglomerado mais estável. O rearranjo da primeira estrutura de transição para a segunda gera um decréscimo de energia de 0.550eV, enquanto que o segundo processo que leva a estrutura estável gera um decréscimo de 0.014eV. A figura 3.20 ilustra o processo de rearranjo do Mg 4 H 5 e as perdas de energia associadas a cada processo. A estrutura linear formada com a adição do quinto hidrogênio tem o mesmo mecanismo de absorção do hidrogênio descrito para o Mg 3 H m linear. Os aglomerados encontrados para o Mg 4 H 5 mostram resultados similares aos obtidos para o Mg 3 H m quando se trata do triângulo formado pelos átomos de magnésio. Como mostra a figura A.3, a adição do hidrogênio na face e no centro 74
77 Figura 3.20: Rearranjo do Mg 4 H 5 e as perdas de energias em cada processo. do triângulo resultam em pontos de cela. Deste ponto em diante, observa-se que as outras estruturas estáveis encontradas apresentam uma substancial diferença de energia em relação à estrutura linear. O sucessivo acréscimo de hidrogênios culmina com a saturação do aglomerado linear assim como o ocorrido para o Mg 3 H m. O efeito da interação entre hidrogênios foi de fundamental importância para o rearranjo da simetria inicial do Mg 4 mais estável. Mesmo sendo a energia de ligação média entre os átomos de magnésio do Mg 4, de 0.313eV, maior que as do Mg 3 e Mg 2, ela ainda é muito inferior a interação entre hidrogênios ou a interação entre magnésio e hidrogênio. Desta maneira, suspeita-se que este rearranjo possa ocorrer para os aglomerados de magnésio maiores cujas energias de ligação média sejam ainda baixas. 75
=1.877 ev Simetria: C S EL(H)=3.186 ev EA=17.346 ev EL(Mg)=0.289 ev EL(MgH)=1.814 ev Simetria: D 2D EL(H)=2.869 ev EA=8.585 ev EL(Mg)=0.440 ev EL(MgH)=1.862 ev Simetria: C 2V EL(H)=2.")
 e a linear é de 0.")
78 EL(H)=1.945 ev EA=3.195 ev EL(Mg)=0.702 ev EL(MgH)=1.123 ev Simetria: C 2V EL(H)=2.636 ev EA= ev EL(Mg)=0.279 ev EL(MgH)=1.267 ev Simetria:C 2V EL(H)=2.826 ev EA=6.021 ev EL(Mg)=0.603 ev EL(MgH)=1.877 ev Simetria: C S EL(H)=3.186 ev EA= ev EL(Mg)=0.289 ev EL(MgH)=1.814 ev Simetria: D 2D EL(H)=2.869 ev EA=8.585 ev EL(Mg)=0.440 ev EL(MgH)=1.862 ev Simetria: C 2V EL(H)=2.147 ev EA= ev EL(Mg)=0.703 ev EL(MgH)= ev Simetria: C 2V EL(H)=2.991 ev EA= ev EL(Mg)=0.334 ev EL(MgH)=1.700 ev Simetria:C 2V EL(H)=3.748 ev EA= ev EL(Mg)=1.247 ev EL(MgH)=2.800 ev Simetria: D 2H Figura 3.21: Estruturas mais estáveis para o Mg 4 H m. 3.6 Mg 5 H m No caso dos aglomerados Mg 5, a diferença de energia entre a geometria mais estável (Fig.3.22a) e a linear é de 0.950eV (Fig.3.22b). Já diferença para a geometria ilustrada na figura 3.22b é de 0.046eV, bem menor que a anterior. Esta pequena diferença sugere que aglomerados Mg 5 com a geometria apresentada em 76
79 3.22b possam formar estruturas estáveis para Mg 5 H m. Sendo assim, usamos as estruturas apresentadas em 3.22a, 3.22b e 3.22c como ponto de partida para os aglomerados Mg 5 H m proveniente de ambos os aglomerados do Mg 5. As estruturas mais estáveis estão listados na figura 3.23 Figura 3.22: Possíveis estruturas para o Mg 5. Novamente usamos os orbitais moleculares do Mg 5 para tentar determinar os sítios preferenciais de ligação do hidrogênio. Estes orbitais estão listados na figura B.12. Os orbitais de fronteira HOMO, LUMO, LUMO+2 e LUMO+3 sugerem que os átomos de hidrogênio podem ligar-se tanto nos átomos quanto nas arestas. A figura A.5 mostra que, de fato, estes sítios resultam em locais de ligação do hidrogênio no Mg 5 H. No caso da aresta da base, o orbital LUMO que poderia mediar a ligação não é tão bem orientado para realizar a ligação do hidrogênio. A figura A.5 mostra ainda que a diferença energética entre as estruturas provenientes dos aglomerados apresentados nas figuras 3.22a e 3.22b aumentam. A diferença que era de apenas 0.046eV entre os aglomerados puros aumenta para 0.169eV após o acréscimo do hidrogênio. A adição do hidrogênio no centro da 77
80 EL(H)= ev EA=3.894 ev Simetria: C 3V EL(H)= ev EA=9.203 ev Simetria: C S EL(H)= ev EA=6.912 ev Simetria: D 3H EL(H)= ev EA= ev Simetria: C S Figura 3.23: Estruturas mais estáveis para o Mg 5 H m. estrutura representa um ponto de cela, como era de se esperar. no Mg 3 H m e no Mg 4 H m. É importante notar que a estrutura linear continua sendo muito desfavorável com relação a mais estável, porém a diferença caí de 0.950eV para 0.788eV. A sua formação pode ser vista como a associação de um átomo de hidrogênio ao Mg 4 H linear ou da adição de um Mg 2 ao Mg 3 H. As energias necessárias para tais processos são ev e 0.522eV, respectivamente. Mesmo assim, a grande diferença energética sugere que esta geometria não seja propícia a se formar. A adição do segundo hidrogênio traz interessantes resultados. A estrutura mais estável continua baseada no isômero da figura 3.22a e a diferença de energia entre as outras estruturas é relativamente grande. A comparação das distribuições eletrônicas do Mg 5 e do Mg 5 H 2 apresentadas na figura 3.24 mostra que a influência 78
81 Figura 3.24: Distribuição eletrônica no a) Mg 5 mais estável; b) Mg 5 H 2 mais estável. da interação entre o par de átomos de hidrogênio diminui o comprimento das ligações, gerando assim uma estrutura mais compacta. Além disso, podemos observar na figura A.5 a existência de uma estrutura com um átomo de hidrogênio em seu interior. Esta estrutura é extremamente desfavorável do ponto de vista energético e sua estabilidade se deve à interação entre os átomos de hidrogênio. Outro fato marcante é alta energia da estrutura linear, que difere 0.939eV da estrutura mais estável. Para o Mg 5 H 3, a estrutura mais estável é a proveniente do Mg 5 ilustrado em Sua geometria, ilustrada na figura 3.23, mostra que a interação entre os átomos de hidrogênio está novamente relacionada com a estabilidade da estrutura. A diferença de energia entre o aglomerado mais estável gerada pelo Mg 5 ilustrado na figura 3.22a é de 0.221eV, um valor razoável. O fato interessante para estes aglomerados é a grande estabilização do caso linear, que passa a diferir apenas 79
82 0.291eV do Mg 5 H 3 mais estável. A inserção do hidrogênio no centro do tetraedro resulta em um ponto de cela. O Mg 5 H 4 mais estável continua baseado na estrutura mostrada na figura 3.22b. Entretanto, a diferença de energia entre a estrutura gerada a partir da estrutura 3.22a é de 0.024eV. Já a estrutura linear tem uma diferença razoável de 0.407eV. Nota-se ainda a presença de uma estrutura onde os átomos de magnésio se encontram coplanares entre si com uma diferença de energia de 0.193eV. No caso dos aglomerados Mg 5 H m, não se observou um rearranjo voluntário dos átomos de magnésio com a adição de átomos de hidrogênio como no Mg 4 H m. As diferentes estruturas foram avaliadas separadamente, sem serem quebradas. Mais uma vez, a interação entre os hidrogênios se mostrou muito importante para a estabilidade das estruturas. A evolução da diferença de energia entre as estruturas mais estáveis e as lineares leva a crer que para uma maior concentração de átomos de hidrogênio a estrutura linear pode vir a se tornar a mais estável. 3.7 Mg 6 H m Na figura 3.25 apresentamos os dois isômeros mais estáveis para os aglomerados Mg 6. Ambas as estruturas são obtidas a partir da adição de um átomo de magnésio na aresta do aglomerado mais estável Mg 5 (Fig. 3.25a) ou em uma de suas faces (Fig. 3.25b). Como a diferença de energia entre as duas estruturas 80
83 é de 0.015eV, podemos esperar que os aglomerados Mg 6 H m formados a partir de ambas as estruturas sejam competitivas entre si. Desta forma, procederemos com cálculos para as duas estruturas paralelamente. A estrutura linear do Mg 6 difere 1.118eV da mais estável. Figura 3.25: a) Estrutura mais estável do Mg 6 ; b) Segundo isômero do Mg 6. As estruturas mais estáveis estão listadas na figura Podemos perceber desta figura que os Mg 6 H m mais estáveis provém do Mg 6 mais estável até a adição do quarto hidrogênio, quando a estrutura do segundo isômero do Mg 6 é encontrada. Ao se comparar com o caso do Mg 5, nota-se que a mudança da geometria do Mg 5 que gera a estrutura mais estável ocorreu para o Mg 5 H 3. A comparação entre o Mg 6 H gerado a partir do primeiro e do segundo isômero do Mg 6, listados na figura A.7, mostra uma diferença energética de 0.209eV, um valor muito maior do que a diferença de 0.015eV entre os aglomerados puros. O primeiro ponto de cela listado em A.7 é um estado de transição entre os Mg 6 H 81



84 EL(H)= ev EA=4.516 ev Simetria: C S EL(H)= ev EA=9.907 ev Simetria: C S EL(H)= ev EA=7.350 ev Simetria: C S EL(H)= ev EA= ev Simetria: C 2V Figura 3.26: Estruturas mais estáveis para o Mg 6 H m. provenientes de ambas as geometrias do Mg 6. Outro fato importante é que a inserção do hidrogênio no centro dos aglomerados resultou em pontos de cela, como constatado nos aglomerados menores. Nota-se que a discrepância entre o Mg 6 H linear e o mais estável aumenta em relação aos aglomerados puros, uma vez que a diferença de energia aumenta de 1.118eV para 1.121eV. Sendo sua ocorrência, portanto, questionável. O acréscimo do segundo hidrogênio no Mg 6 H mostra que a diferença entre as estruturas formadas pelos dois Mg 6 iniciais cai. A figura mostra que o Mg 6 H 2 baseado no segundo isômero do Mg 6 mais estável difere de apenas 0.129eV do Mg 6 H 2 mais estável. A diferença energética do Mg 6 H 2 linear também cai suavemente para 1.102eV, mas mesmo assim ainda é muito desfavorável energeticamente. A sua formação é duvidosa devido a essa discrepância energética, assim como para 82
85 o Mg 5 H. No Mg 6 H 3, apresentado na figura A.8, essas diferenças são novamente reduzidas até que pela adição do quarto hidrogênio a estrutura baseada no segundo isômero do Mg 6 passa a ser a mais estável. Mesmo assim, as estruturas oriundas do Mg 6 mais estável apresentam energias praticamente iguais à do Mg 6 H 4 mais estável. Existem três estruturas que diferem por menos de 0.100eV, sendo que a mais próxima difere apenas 0.024eV. É importante observar que nesse processo a diferença de energia entre os casos mais estáveis e os lineares cai para 0.695eV com a adição do terceiro hidrogênio, e para 0.672eV com o quarto hidrogênio. Assim como no Mg 5 H m, esta diferença de energia decresce com o aumento da concentração de átomos de hidrogênio. Isso mostra que para uma maior quantidade de hidrogênio, a estrutura linear possa ocorrer. 3.8 Considerações Finais A comparação da energia de ligação do primeiro e do segundo átomo de hidrogênio nos aglomerados Mg n é ilustrada na figura Estas energias foram obtidas a partir dos resultados apresentados neste trabalho e de cálculos preliminares isolados para os aglomerados Mg 7, Mg 9, Mg 13 e Mg 18 que não foram a- presentados. Desta figura, podemos especular acerca do comportamento da ligação 83
86 dos hidrogênios em aglomerados maiores. 3,25 3,00 Energia de Ligação (ev) 2,75 2,50 2,25 2,00 1,75 1, número de magnésios (n) Figura 3.27: A linha sólida representa a energia de ligação do primeiro hidrogênio e a pontilhada representa a energia de ligação do segundo hidrogênio. A forte interação entre o hidrogênio e metais pode explicar o aumento da energia de ligação do primeiro hidrogênio apresentado em Como mencionado anteriormente, há indícios de uma transição dos aglomerados de magnésio para uma fase metálica em função do seu aumento. Caso isso aconteça de fato, é de se esperar o suposto crescimento da energia de ligação do primeiro hidrogênio observado. A suposta diminuição da energia de ligação do segundo hidrogênio leva a crer que a interação entre o par de hidrogênios seja atenuada devido ao aumento de possíveis posições para o segundo hidrogênio e o aumento da estabilidade dos 84
87 aglomerados Mg n, como mostra a figura 3.1. Isto indica que um rearranjo do aglomerado Mg n causado por uma pequena concentração de hidrogênio seja pouco provável. As figuras 3.28 e 3.29 mostram a evolução do caráter-p dos aglomerados Mg n H m em função da quantidade de átomos de hidrogênio. Nota-se que com a sucessiva adição de hidrogênio nos aglomerados Mg n puros, os valores do caráterp dos Mg n H m tendem a variar menos para estruturas com um maior número de átomos de magnésio. Isto sugere que as ligações entre os magnésio são menos alteradas pelos primeiros hidrogênio adicionados às estruturas maiores. Estas especulações levam a crer que os maiores aglomerados são bons candidatos para o armazenamento de hidrogênio. É importante ressaltar que esta afirmação é embasada em cálculos preliminares e por isso ela deve ser recebida com cuidado. Para prover o devido lastro a tal informação, são necessários mais estudos acerca desses aglomerados maiores. É interessante notar também que as diferenças entre as estruturas mais estáveis Mg n e seus isômeros lineares parece aumentar em função do crescimento dos aglomerados. A adição dos primeiros hidrogênios em ambas os aglomerados parece diminuir um pouco essa discrepância. Mesmo assim, a diferença continua tão grande que a probabilidade de se encontrar estruturas Mg n H m lineares com uma pequena concentração de hidrogênio deve ser pequena para Mg n formado por muitos átomos. A ocorrência dessas estruturas lineares com maior número 85
88 0,3 0,2 Mg 6 H m 0,1 0,3 0,2 Mg 5 H m 0,1 0,3 Caráter-p 0,2 0,1 Mg 4 H m 0,3 0,2 Mg 3 H m 0,1 0,3 0,2 Mg 2 H m 0, número de hidrogênios (m) Figura 3.28: Caráter-p dos aglomerados Mg n H m para m = 1,.., 6. de magnésios deve ser mais propícia para aglomerados perto da saturação, onde a diferença de energia deve ser menor ou ela possa até ser a estrutura mais estável. 86
89 0,5 0,4 Mg 18 H m 0,3 0,5 Caráter-p 0,4 0,3 Mg 13 H m 0,5 0,4 Mg 9 H m 0,3 0,3 0,2 Mg 7 H m 0, número de hidrogênios (m) Figura 3.29: Caráter-p dos aglomerados Mg n H m para m = 7, 9, 13,
90 Capítulo 4 Conclusão Os resultados obtidos neste trabalho mostram que a forma preferencial de armazenamento do hidrogênio é a atômica. A natureza de sua ligação aos aglomerados de magnésio é covalente. Observamos que a superfície externa das estruturas é mais propícia para o armazenamento do hidrogênio ao se adicioná-lo à diversos sítios dos aglomerados de magnésio, em especial as arestas e os átomos de magnésio. O centro das estruturas não se mostrou um local favorável ao armazenamento do hidrogênio. O sucessivo acréscimo dos átomos de hidrogênio mostrou a existência de um ponto de saturação do aglomerado de magnésio para os aglomerados lineares. A partir deste ponto, a adição de um novo hidrogênio gera uma molécula H 2 que se liga fracamente ao aglomerado Mg n H m 2. Nestes aglomerados lineares saturados, a massa do hidrogênio armazenado corresponde a 7.7% da massa total do Mg n H m. 88
91 O procedimento empregado neste trabalho para a determinação das estruturas Mg n H m mais estável não leva em consideração as reações as quais os aglomerados Mg n H m estão sujeitos. O estudo destas reações fornecerá informações sobre a viabilidade de se obter as diversas estruturas aqui encontradas, indicando assim as estruturas mais prováveis de se formarem com o acrescimo dos átomos de hidrogênio. Outra possibilidade para estudos futuros é o uso de catalisadores para facilitar a extração do hidrogênio nos aglomerados Mg n H m ou da dopagem dos aglomerados Mg n para promover o aumento de sua estabilidade. O uso de metais pesados em estruturas cristalinas de magnésio[26], como o cobre ou o níquel, pode vir a estabilizar ainda mais os aglomerados Mg n. 89
92 Apêndice A Estruturas Estáveis e Pontos de Cela para os Aglomerados Mg n H m 90




93 Mg 2 H Mg 2 H 2 Mg 2 H 3 Mg 2 H 4 PC - C 2V E=0.184 ev PC C 2V E=0.006 ev ML C 2V E=0.602 ev ML C 3V E=0.982 ev ML D H E=0.644 ev ML C 2V E=0.241 ev PC C 2V E=1.018 ev ML D 2H E=0.441 ev Figura A.1: Outras estruturas de maior energia para o Mg 2 H m, onde ML representa um mínimo local e PC um ponto de cela. 91





94 Mg 3H Mg 3H 2 Mg 3H 3 Mg 3H 4 Mg 3H 5 Mg 3H 6 ML C 2V E=0.042 ev ML C 2V E=0.010 ev ML C 2V E=0.166 ev ML C 2V E=0.233 ev ML C 2V E=0.164 ev ML D 3H E=0.144 ev ML C 2V E=0.061 ev ML C 2V E=0.132 ev ML C S E=0.478 ev ML C S E=0.326 ev ML C S E=0.321 ev ML C 2V E=0.196 ev PC D 3H E=0.488 ev ML C S E=0.136 ev ML C S E=0.481 ev ML C 2V E=0.576 ev ML C S E=0.568 ev ML C 2V E=0.314 ev ML C S E=0.653 ev ML C 3V E=0.893 ev ML C 2V E=0.765 ev ML C 2V E=0.322 ev PC C 2V E=0.738 ev PC C S E=0.773 ev ML C 2V E=0.374 ev PC C 2V E=0.401 ev PC C 2V E=0.959 ev Figura A.2: Outras estruturas de maior energia para o Mg 3 H m, onde ML representa um mínimo local e PC um ponto de cela. 92









95 Mg 4H Mg 4H 2 Mg 4H 3 Mg 4H 4 ML C 3V E=0.018 ev ML C 2V E=0.261 ev PC C S E=0.204 ev ML C S E=0.119 ev ML C 3V E=0.029 ev ML C S E=0.275 ev ML Cs E=0.211 ev ML C S E=0.138 ev ML C V E=0.387 ev ML D H E=0.326 ev ML C 2V E=0.454 ev ML C 2V E=0.248 ev PC C 2V E=0.312 ev ML C 2V E=0.541 ev PC C 3V E=1.271 ev PC C S E=0.453 ev PC C 2V E=0.591 ev ML C 3V E=1.743 ev ML C 2 E=0.477 ev PC C 2V E=0.785 ev PC C 2V E=0.477 ev PC C 2V E=1.047 ev Figura A.3: Outras estruturas de maior energia para o Mg 4 H m, m 4, onde ML representa um mínimo local e PC um ponto de cela. 93
96 Mg 4H 5 Mg 4H 6 Mg 4H 7 PC C 2V E=0.014 ev ML C 2V E=1.961 ev ML C S E=0.449 ev ML C S E=1.006 ev ML C S E=0.225 ev PC C 2V E=2.117 ev PC C S E=0.717 ev ML C S E=0.258 ev PC C S E=0.329 ev PC C 2V E=0.564 ev PC C S E=0.683 ev PC C 2V E=0.730 ev Figura A.4: Outras estruturas de maior energia para o Mg 4 H m, m > 4, onde ML representa um mínimo local e PC um ponto de cela. 94











97 Mg 5H Mg 5H 2 ML C S E=0.060 ev PC C 4V E=0.350 ev ML C S E=0.224 ev ML C 2V E=0.167 ev PC D 3H E=0.380 ev ML C 2V E=0.286 ev ML C 3V E=0.169 ev PC C 2V E=0.448 ev ML C 3V E=0.487 ev PC C 2V E=0.186 ev ML C V E=0.788 ev ML C 2V E=0.489 ev PC C 2V E=0.240 ev ML C S E=0.491 ev ML C 3V E=0.320 ev ML C 2V E=0.819 ev ML C 2V E=0.348 ev ML D H E=0.939 ev Figura A.5: Outras estruturas de maior energia para o Mg 5 H m, m 2, onde ML representa um mínimo local e PC um ponto de cela. 95
98 Mg 5H 3 Mg 5H 4 ML C S E=0.221 ev ML C 2V E=0.505 ev ML C S E=0.024 ev ML C 2V E=0.291 ev PC C 2V E=0.585 ev ML C S E=0.131 ev ML C S E=0.331 ev ML C 2V E=0.193 ev ML C 1 E=0.373 ev PC C S E=0.220 ev PC C S E=0.385 ev ML C 2V E=0.407 ev PC C S E=0.392 ev PC C 2V E=0.568 ev ML C 2V E=0.441 ev ML C 2V E=0.787 ev Figura A.6: Outras estruturas de maior energia para o Mg 5 H m, m > 2, onde ML representa um mínimo local e PC um ponto de cela. 96

















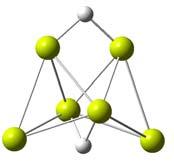




99 Mg 6H Mg 6H 2 ET C 2V E=0.170 ev ET C S E=0.565 ev ML C 1 E=0.107 ev ML C 2 E=0.240eV ML C S E=0.209eV ML C S E=0.573 ev ML C S E=0.117eV ET C 2V E=0.253 ev ET C S E=0.270 ev ET C 2V E=0.653 ev ML C S E=0.129 ev ML C S E=0.353 ev ML C S E=0.344 ev ET C 2V E=0.689 ev ML C S E=0.136 ev ML C S E=0.572 ev ML C 1 E=0.347 ev ET C 2V E=0.790 ev ML C S E=0.178 ev ET C 2V E=0.576 ev ML C S E=0.417 ev ML C V E=1.121 ev ML C S E=0.191 ev ML D H E=1.102eV ML C S E=0.494 ev ML C S E=0.236 ev Figura A.7: Outras estruturas de maior energia para o Mg 6 H m, m 2, onde ML representa um mínimo local e PC um ponto de cela. 97





100 Mg 6 H 3 Mg 6 H 4 ML C S E=0.132 ev ML C 1 E=0.024 ev ML C S E=0.149 ev ML C S E=0.063 ev ML C S E=0.150 ev ML C S E=0.080 ev ML C S E=0.193 ev ML C 1 E=0.119 ev ML C 1 E=0.386 ev ML C S E=0.123 ev ML C 2V E=0.695 ev ML C 2V E=0.672 ev Figura A.8: Outras estruturas de maior energia para o Mg 6 H m, m > 2, onde ML representa um mínimo local e PC um ponto de cela. 98
101 Apêndice B Orbitais Moleculares dos Aglomerados Mg n H m 99



102 Mg MgH Lumo+2 Lumo+1 Lumo Homo Homo-1 Figura B.1: Orbitais atômicos do magnésio e moleculares do MgH. 100



103 MgH 2 LUMO+2 LUMO+1 LUMO HOMO HOMO-1 Figura B.2: Orbitais moleculares do MgH






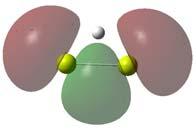









104 Mg 2 Mg 2 H - átomo Mg 2 H - aresta LUMO+3 LUMO+2 LUMO+1 LUMO HOMO HOMO-1 HOMO-2 Figura B.3: Orbitais moleculares do Mg 2 e Mg 2 H para duas geometrias. 102





105 Mg 2 H 2 - linear Mg 2 H 2 - aresta LUMO+3 LUMO+2 LUMO+1 LUMO HOMO HOMO-1 HOMO-2 Figura B.4: Orbitais moleculares do Mg 2 H 2 para duas geometrias. 103










106 Mg 2 H 3 Mg 2 H 3 LUMO+3 LUMO+2 LUMO+1 LUMO HOMO HOMO-1 HOMO-2 Figura B.5: Orbitais moleculares do Mg 2 H 3 para duas geometrias. 104



")




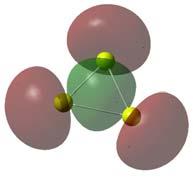













107 Mg 3 Mg 3 - linha Mg 3H - linear Mg 3H - átomo Mg 3H - aresta LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.6: Orbitais moleculares do Mg 3 e do Mg 3 H. 105









108 Mg 3 H 2 - linear Mg 3 H 2 - triangular Mg 3 H 2 linear C 2V Mg 3 H 2 linear C 2V LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.7: Orbitais moleculares do Mg 3 H



























109 Mg 3H 3 - linear Mg 3H 3 - aresta Mg 3H 3 - atomo Mg 3H 3 linear de maior energia Mg 3H 3 face LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.8: Orbitais moleculares do Mg 3 H

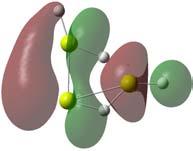











110 Mg 3 H 4 - linear Mg 3 H 4 - triangular LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.9: Orbitais moleculares do Mg 3 H























111 Mg 3H 5 - linear Mg 3H 5 - triangular Mg 3H 5 - faces Mg 3H 5 linear de maior energia LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.10: Orbitais moleculares do Mg 3 H

















112 Mg 4 Mg 4 H - aresta Mg 4 H - átomo LUMO+3 LUMO+2 LUMO+1 LUMO HOMO HOMO-1 HOMO-2 HOMO-3 Figura B.11: Orbitais moleculares do Mg 4 e do Mg 4 H. 110












113 Mg 5 Mg 5 H LUMO+3 (alfa) LUMO+2 (alfa) LUMO+1 (alfa) LUMO (alfa) HOMO (alfa) HOMO-1 (alfa) HOMO-2 (alfa) Figura B.12: Orbitais moleculares do Mg 5 e do Mg 5 H. 111
O Método de Hartree-Fock
 O Método de Hartree-Fock CF740 Tópicos Especiais de Física Atômica e Molecular Cálculos de Estrutura Eletrônica Utilizando Funcionais de Densidade Departamento de Física Universidade Federal do Paraná
O Método de Hartree-Fock CF740 Tópicos Especiais de Física Atômica e Molecular Cálculos de Estrutura Eletrônica Utilizando Funcionais de Densidade Departamento de Física Universidade Federal do Paraná
2 Teoria do Funcional da Densidade
 2 Teoria do Funcional da Densidade A Mecânica Quântica teve seu início com a equação de Schrödinger (1926). Esta equação determina a função de onda quântica de um sistema, seja ele um átomo, uma molécula
2 Teoria do Funcional da Densidade A Mecânica Quântica teve seu início com a equação de Schrödinger (1926). Esta equação determina a função de onda quântica de um sistema, seja ele um átomo, uma molécula
Introdução ao Método de Hartree-Fock
 Introdução ao Método de Hartree-Fock CF352 - Fundamentos de Física Atômica e Molecular Departamento de Física Universidade Federal do Paraná M. H. F. Bettega (UFPR) CF352 1 / 24 Preliminares Aproximação
Introdução ao Método de Hartree-Fock CF352 - Fundamentos de Física Atômica e Molecular Departamento de Física Universidade Federal do Paraná M. H. F. Bettega (UFPR) CF352 1 / 24 Preliminares Aproximação
Operadores e Função de Onda para Muitos Elétrons. Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros
 Operadores e Função de Onda para Muitos Elétrons Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros Livro texto: Modern Quantum Chemistry Introduction to Advanced Elecronic Structure Theory
Operadores e Função de Onda para Muitos Elétrons Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros Livro texto: Modern Quantum Chemistry Introduction to Advanced Elecronic Structure Theory
FF-296: Teoria do Funcional da Densidade I. Ronaldo Rodrigues Pela
 FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos HF Dois elétrons N elétrons Thomas Fermi Átomo de Hélio 1D Energia exata: 3,154 H Vamos resolver este problema usando o método
FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos HF Dois elétrons N elétrons Thomas Fermi Átomo de Hélio 1D Energia exata: 3,154 H Vamos resolver este problema usando o método
NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA
 NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA Prof. Carlos R. A. Lima CAPÍTULO 10 ÁTOMOS COMPLEXOS Primeira Edição junho de 2005 CAPÍTULO 10 ÁTOMOS COMPLEXOS ÍNDICE 10-1- Introdução 10.2- Átomos com mais de um
NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA Prof. Carlos R. A. Lima CAPÍTULO 10 ÁTOMOS COMPLEXOS Primeira Edição junho de 2005 CAPÍTULO 10 ÁTOMOS COMPLEXOS ÍNDICE 10-1- Introdução 10.2- Átomos com mais de um
FF-296: Teoria do Funcional da Densidade I. Ronaldo Rodrigues Pela
 FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos O problema de 1 elétron O princípio variacional Função de onda tentativa Átomo de H unidimensional Íon H2 + unidimensional Equação
FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos O problema de 1 elétron O princípio variacional Função de onda tentativa Átomo de H unidimensional Íon H2 + unidimensional Equação
FF-296: Teoria do Funcional da Densidade I. Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785
 FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Hartree-Fock e Thomas-Fermi Hartree-Fock Caso de N
FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Hartree-Fock e Thomas-Fermi Hartree-Fock Caso de N
FF-296: Teoria do Funcional da Densidade I. Ronaldo Rodrigues Pela
 FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos Teorema de Hohenberg-Kohn Equações de Kohn-Sham Problema de N elétrons Aproximação de Born-Oppenheimer: os núcleos estão totalmente
FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos Teorema de Hohenberg-Kohn Equações de Kohn-Sham Problema de N elétrons Aproximação de Born-Oppenheimer: os núcleos estão totalmente
UMA SIMPLIFICAÇÃO DO MÉTODO HARTREE-FOCK
 UMA SIMPLIFICAÇÃO DO MÉTODO HARTREE-FOCK Lucas Modesto da Costa Instituto de Física - DFGE - USP 8 de outubro de 2008 1 / 39 Slater, J. C. Época Aproximação de Thomas-Fermi 2 / 39 Slater, J. C. Slater,
UMA SIMPLIFICAÇÃO DO MÉTODO HARTREE-FOCK Lucas Modesto da Costa Instituto de Física - DFGE - USP 8 de outubro de 2008 1 / 39 Slater, J. C. Época Aproximação de Thomas-Fermi 2 / 39 Slater, J. C. Slater,
FF-296: Teoria do Funcional da Densidade I. Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785
 FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Hartree-Fock e Thomas-Fermi Teorema de Hohenberg-Kohn
FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Hartree-Fock e Thomas-Fermi Teorema de Hohenberg-Kohn
FF-296: Teoria do Funcional da Densidade I. Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785
 FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Problema de 2 elétrons Férmions Hartree-Fock Troca
FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tema de hoje: Problema de 2 elétrons Férmions Hartree-Fock Troca
Correlação Eletrônica - CI e MP2
 Correlação Eletrônica - CI e MP2 CF740 Tópicos Especiais de Física Atômica e Molecular Cálculos de Estrutura Eletrônica Utilizando Funcionais de Densidade Departamento de Física Universidade Federal do
Correlação Eletrônica - CI e MP2 CF740 Tópicos Especiais de Física Atômica e Molecular Cálculos de Estrutura Eletrônica Utilizando Funcionais de Densidade Departamento de Física Universidade Federal do
Moléculas. Usamos a aproximação de Born- Oppenheimer, que considera os núcleos fixos, apenas o e - se movimenta.
 Moléculas Moléculas é uma coleção de 2 ou mais núcleos e seus elétrons associados, com todas as ligações complexas unidas pelas forças Os tipos mais importantes de ligação molecular são: covalente, iônica
Moléculas Moléculas é uma coleção de 2 ou mais núcleos e seus elétrons associados, com todas as ligações complexas unidas pelas forças Os tipos mais importantes de ligação molecular são: covalente, iônica
Teoria do Funcional de Densidade
 Teoria do Funcional de Densidade Márcio H. F. Bettega Departamento de Física Universidade Federal do Paraná bettega@fisica.ufpr.br M. H. F. Bettega (UFPR) PG Física 1 / 23 Kohn recebendo seu Prêmio Nobel
Teoria do Funcional de Densidade Márcio H. F. Bettega Departamento de Física Universidade Federal do Paraná bettega@fisica.ufpr.br M. H. F. Bettega (UFPR) PG Física 1 / 23 Kohn recebendo seu Prêmio Nobel
Introdução à Física Atômica e Molecular
 4300315 Introdução à Física Atômica e Molecular Átomo de Hélio * : Estado Fundamental II *Referência Principal: D. Vianna, A. Fazzio e S. Canuto, Teoria Quântica de Moléculas e Sólidos Átomo de Hélio Aproximação
4300315 Introdução à Física Atômica e Molecular Átomo de Hélio * : Estado Fundamental II *Referência Principal: D. Vianna, A. Fazzio e S. Canuto, Teoria Quântica de Moléculas e Sólidos Átomo de Hélio Aproximação
Física Moderna II - FNC376
 Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto 1o. Semestre de 2008 1 MQ átomos > < Moléculas moléculas e sólidos núcleos e partículas Moléculas
Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto 1o. Semestre de 2008 1 MQ átomos > < Moléculas moléculas e sólidos núcleos e partículas Moléculas
Átomos polieletrónicos
 Átomos polieletrónicos Química Teórica e Estrutural P.J.S.B. Caridade & U. Miranda 2/12/2013 5/11/2013, Aula 8 Química Teórica & Estrutural (2013) Caridade & Ulises 1 Átomo de hidrogénio O Hamiltoniano
Átomos polieletrónicos Química Teórica e Estrutural P.J.S.B. Caridade & U. Miranda 2/12/2013 5/11/2013, Aula 8 Química Teórica & Estrutural (2013) Caridade & Ulises 1 Átomo de hidrogénio O Hamiltoniano
Data de entrega dos resumos
 Data de entrega dos resumos Primeiro resumo Até 25 de abril de 2018 quarta-feira Segundo resumo Até 20 de junho de 2018 quarta-feira Cap. 3 Terceira parte Teorema de Koopmans Teorema de Brillouin Hamiltoniano
Data de entrega dos resumos Primeiro resumo Até 25 de abril de 2018 quarta-feira Segundo resumo Até 20 de junho de 2018 quarta-feira Cap. 3 Terceira parte Teorema de Koopmans Teorema de Brillouin Hamiltoniano
Operadores e Função de Onda para Muitos Elétrons. Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros
 Operadores e Função de Onda para Muitos Elétrons Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros Aproximação de Hartree-Fock A maior preocupação da química quântica é encontrar e descrever
Operadores e Função de Onda para Muitos Elétrons Introdução à Física Atômica e Molecular UEG Prof. Renato Medeiros Aproximação de Hartree-Fock A maior preocupação da química quântica é encontrar e descrever
Geralmente, fazemos simplificações e desenvolvemos modelos. A matéria é composta por átomos e moléculas
 Disciplina: SiComLíMol 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite tratar quantidades macroscópicas da matéria.
Disciplina: SiComLíMol 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite tratar quantidades macroscópicas da matéria.
NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA
 NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA Prof. Carlos R. A. Lima CAPÍTULO 11 MOLÉCULAS Primeira Edição junho de 2005 CAPÍTULO 11 MOLÉCULAS ÍNDICE 11-1- Introdução 11.2- Ligação por Tunelamento e a Molécula
NOTAS DE AULAS DE ESTRUTURA DA MATÉRIA Prof. Carlos R. A. Lima CAPÍTULO 11 MOLÉCULAS Primeira Edição junho de 2005 CAPÍTULO 11 MOLÉCULAS ÍNDICE 11-1- Introdução 11.2- Ligação por Tunelamento e a Molécula
A Estrutura Eletrônica dos Átomos. Prof. Fernando R. Xavier
 A Estrutura Eletrônica dos Átomos Prof. Fernando R. Xavier UDESC 2015 Estrutura Atômica, Antencedentes... Modelos de Demócrito, Dalton, Thomson, etc 400 a.c. até 1897 d.c. Nascimento da Mecânica Quântica
A Estrutura Eletrônica dos Átomos Prof. Fernando R. Xavier UDESC 2015 Estrutura Atômica, Antencedentes... Modelos de Demócrito, Dalton, Thomson, etc 400 a.c. até 1897 d.c. Nascimento da Mecânica Quântica
Ligações Químicas - I
 Ligações Químicas - I Orbitais atômicos e números quânticos A tabela periódica; propriedades Ligações químicas A ligação iônica Ligação covalente Orbitais moleculares (LCAO) Hibridização Geometrias moleculares
Ligações Químicas - I Orbitais atômicos e números quânticos A tabela periódica; propriedades Ligações químicas A ligação iônica Ligação covalente Orbitais moleculares (LCAO) Hibridização Geometrias moleculares
Átomos e Moléculas. Ligações moleculares. Energia do ion. A molécula de hidrogênio H 2
 Ligações moleculares Átomos e Moléculas Energia do ion H 2 + A molécula de hidrogênio Ligações moleculares Uma molécula é formada por um conjunto de átomos que interagem formando um sistema com energia
Ligações moleculares Átomos e Moléculas Energia do ion H 2 + A molécula de hidrogênio Ligações moleculares Uma molécula é formada por um conjunto de átomos que interagem formando um sistema com energia
Universidade do Estado de Santa Catarina UDESC - CCT Química Geral Profª Fabíola Corrêa viel
 Ligações químicas Universidade do Estado de Santa Catarina UDESC - CCT Química Geral Profª Fabíola Corrêa viel Ligações Químicas Sacarina Vitamina E Amônia Fulereno LIGAÇÕES IÔNICAS Acontece por atração
Ligações químicas Universidade do Estado de Santa Catarina UDESC - CCT Química Geral Profª Fabíola Corrêa viel Ligações Químicas Sacarina Vitamina E Amônia Fulereno LIGAÇÕES IÔNICAS Acontece por atração
Física Moderna II Aula 07
 Universidade de São Paulo Instituto de Física 1 º Semestre de 015 Profa. Márcia de Almeida Rizzutto Oscar Sala sala 0 rizzutto@if.usp.br Física Moderna II Aula 07 Monitor: Gabriel M. de Souza Santos Sala
Universidade de São Paulo Instituto de Física 1 º Semestre de 015 Profa. Márcia de Almeida Rizzutto Oscar Sala sala 0 rizzutto@if.usp.br Física Moderna II Aula 07 Monitor: Gabriel M. de Souza Santos Sala
1 Ψ S. bósons estatística de Bose: n(e) = e α e E/kT 1 1 Ψ A. férmions estatística de Fermi: n(e) = e α e E/kT +1
 = e α e E/kT 1 1 Ψ A. férmions estatística de Fermi: n(e) = e α e E/kT +1") Estatística Quântica Boltzmann: n(e) = 1 e α e E/kT Indistinguibilidade Ψ S ou Ψ A 1 Ψ S bósons estatística de Bose: n(e) = e α e E/kT 1 1 Ψ A férmions estatística de Fermi: n(e) = e α e E/kT +1 1 Distribuição
Estatística Quântica Boltzmann: n(e) = 1 e α e E/kT Indistinguibilidade Ψ S ou Ψ A 1 Ψ S bósons estatística de Bose: n(e) = e α e E/kT 1 1 Ψ A férmions estatística de Fermi: n(e) = e α e E/kT +1 1 Distribuição
Física de Semicondutores. Aula 9 DEFEITOS EM SEMICONDUTORES
 Física de Semicondutores Aula 9 DEFEITOS EM SEMICONDUTORES Defeitos permitem controlar o comportamento elétrico e/ou óptico dos materiais e estruturas semicondutoras; tornam possível a imensa variedade
Física de Semicondutores Aula 9 DEFEITOS EM SEMICONDUTORES Defeitos permitem controlar o comportamento elétrico e/ou óptico dos materiais e estruturas semicondutoras; tornam possível a imensa variedade
Física da Matéria Condensada - GFI 04129
 Física da Matéria Condensada - GFI 04129 Antonio T. Costa I semestre, 2007 Programa 1 A estrutura eletrônica de sólidos cristalinos a O que é um sólido cristalino b Comportamento do elétron num sólido
Física da Matéria Condensada - GFI 04129 Antonio T. Costa I semestre, 2007 Programa 1 A estrutura eletrônica de sólidos cristalinos a O que é um sólido cristalino b Comportamento do elétron num sólido
Disciplina: FGE5748 Simulação Computacional de Líquidos Moleculares 1
 Disciplina: FGE5748 Simulação Computacional de Líquidos Moleculares 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite
Disciplina: FGE5748 Simulação Computacional de Líquidos Moleculares 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite
Maxwell Severo da COSTA; Freddy Fernandes GUIMARÃES
 Comparação entre a aproximação Z+1 e o cálculo de buraco de camada real no estudo da dinâmica nuclear dos isômeros do dicloroetileno induzida pela foto-ionização dos elétrons da camada K do cloro (ClK)
Comparação entre a aproximação Z+1 e o cálculo de buraco de camada real no estudo da dinâmica nuclear dos isômeros do dicloroetileno induzida pela foto-ionização dos elétrons da camada K do cloro (ClK)
Cleber Fabiano do Nascimento Marchiori
 Cleber Fabiano do Nascimento Marchiori Programa de Pós -Graduação em Física - Departamento de Física- UFPR Estrutura Eletrônica de Moléculas Orgânicas Curitiba Janeiro, 2014 Sumário LISTA DE FIGURAS 1
Cleber Fabiano do Nascimento Marchiori Programa de Pós -Graduação em Física - Departamento de Física- UFPR Estrutura Eletrônica de Moléculas Orgânicas Curitiba Janeiro, 2014 Sumário LISTA DE FIGURAS 1
PPGQTA. Prof. MGM D Oca
 PPGQTA Prof. Polarizabilidade: Dureza e Moleza A polarizabilidade está relacionada ao tamanho do átomo e da capacidade deste estabilizar elétrons na nuvem eletrônica, esta matematicamente correlacionada
PPGQTA Prof. Polarizabilidade: Dureza e Moleza A polarizabilidade está relacionada ao tamanho do átomo e da capacidade deste estabilizar elétrons na nuvem eletrônica, esta matematicamente correlacionada
PRINCÍPIO BÁSICO: Isótopo Estável + Partícula incidente (n, p, a, g ) Isótopo Radioativo + Partículas emitidas. Medida das radiações emitidas
 Isótopo Radioativo + Partículas emitidas. Medida das radiações emitidas") Radioquímica PRINCÍPIO BÁSICO: Isótopo Estável + Partícula incidente (n, p, a, g ) Isótopo Radioativo + Partículas emitidas (n, p, a, g ) Medida das radiações emitidas O Núcleo Atómico Importância de A
Radioquímica PRINCÍPIO BÁSICO: Isótopo Estável + Partícula incidente (n, p, a, g ) Isótopo Radioativo + Partículas emitidas (n, p, a, g ) Medida das radiações emitidas O Núcleo Atómico Importância de A
5.4.1 Estrutura de bandas para uma única bicamada-rashba com inclusão de tunelamento quântico entre estados helicoidais e não helicoidais
 5.4 Inclusão do tunelamento quântico e hopping de elétrons entre estados helicoidais e não hélicoidais71 h + R 0 D 1 D 12 0 0 0 0... 0 h + D 12 D 2 0 0 0 0... D 1 D 12 h R 0 tz 1 tz 21 0 0... D 12 D 2
5.4 Inclusão do tunelamento quântico e hopping de elétrons entre estados helicoidais e não hélicoidais71 h + R 0 D 1 D 12 0 0 0 0... 0 h + D 12 D 2 0 0 0 0... D 1 D 12 h R 0 tz 1 tz 21 0 0... D 12 D 2
FF-296: Teoria do Funcional da Densidade I. Ronaldo Rodrigues Pela
 FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos Teorema de Hellman-Feynman Conexão adiabática Buraco de XC Sucesso do LDA Estados excitados Teorema de Koopmans Teor. de Hellmann-Feynmann
FF-296: Teoria do Funcional da Densidade I Ronaldo Rodrigues Pela Tópicos Teorema de Hellman-Feynman Conexão adiabática Buraco de XC Sucesso do LDA Estados excitados Teorema de Koopmans Teor. de Hellmann-Feynmann
Cap. 41 -Condução de eletricidade em sólidos
 Cap. 41 -Condução de eletricidade em sólidos Propriedades elétricas dos sólidos; Níveis de energia em um sólido cristalino: Átomo; Molécula; Sólido. Estrutura eletrônica e condução: Isolantes (T = 0);
Cap. 41 -Condução de eletricidade em sólidos Propriedades elétricas dos sólidos; Níveis de energia em um sólido cristalino: Átomo; Molécula; Sólido. Estrutura eletrônica e condução: Isolantes (T = 0);
CÁLCULO DA ESTRUTURA ELETRÔNICA DO ÓXIDO DE GRAFENO
 CÁLCULO DA ESTRUTURA ELETRÔNICA DO ÓXIDO DE GRAFENO Nome do Aluno 1 ; Nome do Orientador 2 ; Nome do Co-orientador 3 (apenas quando estiver oficialmente cadastrado como co-orientador junto ao PIBIC/PIVIC)
CÁLCULO DA ESTRUTURA ELETRÔNICA DO ÓXIDO DE GRAFENO Nome do Aluno 1 ; Nome do Orientador 2 ; Nome do Co-orientador 3 (apenas quando estiver oficialmente cadastrado como co-orientador junto ao PIBIC/PIVIC)
Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais
 Ligações Químicas Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais Ligação iônica Transferência de elétrons de um átomo para outro Íons de cargas opostas Forças
Ligações Químicas Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais Ligação iônica Transferência de elétrons de um átomo para outro Íons de cargas opostas Forças
A eq. de Schrödinger em coordenadas esféricas
 A eq. de Schrödinger em coordenadas esféricas A autofunção espacial, ψ, e a energia, E, são determinadas pela solução da equação independente do tempo: Separação de variáveis Solução do tipo: Que leva
A eq. de Schrödinger em coordenadas esféricas A autofunção espacial, ψ, e a energia, E, são determinadas pela solução da equação independente do tempo: Separação de variáveis Solução do tipo: Que leva
Ligação Covalente. O íon molecular H 2
 O íon molecular H 2 + Dois núcleos de hidrogênio estão ligados por um único elétron O que acontece à medida que os núcleos se aproximam? 4 O íon molecular H 2 + Dois núcleos de hidrogênio estão ligados
O íon molecular H 2 + Dois núcleos de hidrogênio estão ligados por um único elétron O que acontece à medida que os núcleos se aproximam? 4 O íon molecular H 2 + Dois núcleos de hidrogênio estão ligados
A Tabela Periódica pode ser um guia para a ordem na qual os orbitais são preenchidos...
 Aula 02 - Tabela Periódica TABELA PERIÓDICA e Lothar Meyer A Tabela Periódica pode ser um guia para a ordem na qual os orbitais são preenchidos... Pode-se escrever a distribuição eletrônica de um elemento
Aula 02 - Tabela Periódica TABELA PERIÓDICA e Lothar Meyer A Tabela Periódica pode ser um guia para a ordem na qual os orbitais são preenchidos... Pode-se escrever a distribuição eletrônica de um elemento
Química Teórica e Estrutural: Aula 4a
 Química Teórica e Estrutural: Aula 4a P.J.S.B. Caridade & U. Miranda October 16, 2012 Partícula na caixa de potencial: Exemplos práticos Caridade & Miranda TP: aula 4a 2 Postulados da Mecânica Quântica
Química Teórica e Estrutural: Aula 4a P.J.S.B. Caridade & U. Miranda October 16, 2012 Partícula na caixa de potencial: Exemplos práticos Caridade & Miranda TP: aula 4a 2 Postulados da Mecânica Quântica
LIGAÇÃO COVAL COV AL NT
 LIGAÇÃO COVALENTE INTRODUÇÃO Resulta do compartilhamento de elétrons. Ex: H 2 Os dois átomos de hidrogênio se aproximam devido à força de atração que depois de determinada distância diminuem surgindo as
LIGAÇÃO COVALENTE INTRODUÇÃO Resulta do compartilhamento de elétrons. Ex: H 2 Os dois átomos de hidrogênio se aproximam devido à força de atração que depois de determinada distância diminuem surgindo as
Seminário. Teoria de Ligação de Valência - TLV UMA ABORDAGEM MECÂNICO QUÂNTICA DAS LIGAÇÕES COVALENTES
 Seminário Teoria de Ligação de Valência - TLV UMA ABORDAGEM MECÂNICO QUÂNTICA DAS LIGAÇÕES COVALENTES Autor: Marcelo Alves de Souza Mestrando UFABC 2015 Disciplina Química Integrada 1 O modelo de repulsão
Seminário Teoria de Ligação de Valência - TLV UMA ABORDAGEM MECÂNICO QUÂNTICA DAS LIGAÇÕES COVALENTES Autor: Marcelo Alves de Souza Mestrando UFABC 2015 Disciplina Química Integrada 1 O modelo de repulsão
Física Moderna 2. Aula 9. Moléculas. Tipos de ligações. Íon H2 + Iônica Covalente Outras: ponte de hidrogênio van der Waals
 Física Moderna 2 Aula 9 Moléculas Tipos de ligações Iônica Covalente Outras: Íon H2 + ponte de hidrogênio van der Waals 1 Moléculas Uma molécula é um arranjo estável de dois ou mais átomos. Por estável
Física Moderna 2 Aula 9 Moléculas Tipos de ligações Iônica Covalente Outras: Íon H2 + ponte de hidrogênio van der Waals 1 Moléculas Uma molécula é um arranjo estável de dois ou mais átomos. Por estável
Disciplina: SiComLíMol 1
 Disciplina: SiComLíMol 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite tratar quantidades macroscópicas da matéria.
Disciplina: SiComLíMol 1 A matéria é composta por átomos e moléculas Geralmente, fazemos simplificações e desenvolvemos modelos O estudo teórico não permite tratar quantidades macroscópicas da matéria.
Definições de Estabilidade
 Radioquímica Definições de Estabilidade 1. Não se deteta radioatividade. Não há transformação em outro nuclídeo.. Sistema nuclear é estável em relação a outro quando a diferença de energia é negativa:
Radioquímica Definições de Estabilidade 1. Não se deteta radioatividade. Não há transformação em outro nuclídeo.. Sistema nuclear é estável em relação a outro quando a diferença de energia é negativa:
Estrutura Atômica. Química Quântica Prof a. Dr a. Carla Dalmolin. Átomos Polieletrônicos
 Estrutura Atômica Química Quântica Prof a. Dr a. Carla Dalmolin Átomos Polieletrônicos Átomos Polieletrônicos Átomos que possuem mais de 1 elétron A Eq. de Schrödinger pode ser resolvida exatamente apenas
Estrutura Atômica Química Quântica Prof a. Dr a. Carla Dalmolin Átomos Polieletrônicos Átomos Polieletrônicos Átomos que possuem mais de 1 elétron A Eq. de Schrödinger pode ser resolvida exatamente apenas
Mecânica Quântica e Indiscernibilidade
 Mecânica Quântica e Indiscernibilidade t ou ou?? Mecânica clássica Partículas discerníveis ( A, A ) ψ ( A A ) ψ =, Mecânica quântica Partículas indiscerníveis ( A, A ) ψ ( A A ) ψ = ψ, ou = ( A, A ) ψ
Mecânica Quântica e Indiscernibilidade t ou ou?? Mecânica clássica Partículas discerníveis ( A, A ) ψ ( A A ) ψ =, Mecânica quântica Partículas indiscerníveis ( A, A ) ψ ( A A ) ψ = ψ, ou = ( A, A ) ψ
Introdução à Física Atômica e Molecular
 4005 Introdução à Física Atômica e Molecular Átomos Multieletrônicos Referências: D. Vianna, A. Fazzio e S. Canuto, Teoria Quântica de Moléculas e Sólidos. P. Atkins e R. Friedman, Molecular Quantum Mechanics,
4005 Introdução à Física Atômica e Molecular Átomos Multieletrônicos Referências: D. Vianna, A. Fazzio e S. Canuto, Teoria Quântica de Moléculas e Sólidos. P. Atkins e R. Friedman, Molecular Quantum Mechanics,
Física Moderna II - FNC376
 Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto o. Semestre de 008 Conteúdo (P) Cap. 7, 8 e 9 Eisberg (/ do 9.7), Cap. 7 do Tipler, Cap. 7 e parte
Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto o. Semestre de 008 Conteúdo (P) Cap. 7, 8 e 9 Eisberg (/ do 9.7), Cap. 7 do Tipler, Cap. 7 e parte
ESTUDO DE PROPRIEDADES ELETRÔNICAS DE NANOFIOS SEMICONDUTORES DE InAs
 UNIVERSIDADE FEDERAL DE UBERLÂNDIA 4ª Semana do Servidor e 5ª Semana Acadêmica 2008 UFU 30 anos ESTUDO DE PROPRIEDADES ELETRÔNICAS DE NANOFIOS SEMICONDUTORES DE InAs Erika Nascimento Lima 1 Universidade
UNIVERSIDADE FEDERAL DE UBERLÂNDIA 4ª Semana do Servidor e 5ª Semana Acadêmica 2008 UFU 30 anos ESTUDO DE PROPRIEDADES ELETRÔNICAS DE NANOFIOS SEMICONDUTORES DE InAs Erika Nascimento Lima 1 Universidade
INTRODUÇÃO DOS MÉTODOS COMPUTACIONAIS DE PRIMEIROS PRINCÍPIOS PARA O ESTUDO TEÓRICO DE MATERIAIS
 1 INTRODUÇÃO DOS MÉTODOS COMPUTACIONAIS DE PRIMEIROS PRINCÍPIOS PARA O ESTUDO TEÓRICO DE MATERIAIS Fábio Rogério S. Nascimento 1 - Unifesspa Jeânderson de Melo Dantas 2 Unifesspa Agência Financiadora:
1 INTRODUÇÃO DOS MÉTODOS COMPUTACIONAIS DE PRIMEIROS PRINCÍPIOS PARA O ESTUDO TEÓRICO DE MATERIAIS Fábio Rogério S. Nascimento 1 - Unifesspa Jeânderson de Melo Dantas 2 Unifesspa Agência Financiadora:
Estrutura Hiperfina e Efeito Zeeman para Muônio e Positrônio
 Estrutura Hiperfina e Efeito Zeeman para Muônio e Positrônio Armando Valter Felicio Zuffi 05 de dezembro de 014 Resumo Neste trabalho, serão exploradas algumas correções da hamiltoniana do átomo de hidrogênio.
Estrutura Hiperfina e Efeito Zeeman para Muônio e Positrônio Armando Valter Felicio Zuffi 05 de dezembro de 014 Resumo Neste trabalho, serão exploradas algumas correções da hamiltoniana do átomo de hidrogênio.
2.2.1 Efeito Hall e Magnetoresistência Condutividade Elétrica AC Corrente Elétrica em um Campo Magnético
 Conteúdo 1 Revisão de Física Moderna 1 1.1 Equação de Schrödinger; Autoestados e Valores Esperados.. 1 1.2 O Poço de Potencial Innito:Quantização da Energia.............................. 7 1.3 O Oscilador
Conteúdo 1 Revisão de Física Moderna 1 1.1 Equação de Schrödinger; Autoestados e Valores Esperados.. 1 1.2 O Poço de Potencial Innito:Quantização da Energia.............................. 7 1.3 O Oscilador
ÁTOMO: núcleo muito pequeno composto por prótons e nêutrons, que é circundado por elétrons em movimento;
 1.1 CONCEITOS FUNDAMENTAIS ÁTOMO: núcleo muito pequeno composto por prótons e nêutrons, que é circundado por elétrons em movimento; Elétrons e prótons são eletricamente carregados: 1,60 x 10-19 C; Elétrons:
1.1 CONCEITOS FUNDAMENTAIS ÁTOMO: núcleo muito pequeno composto por prótons e nêutrons, que é circundado por elétrons em movimento; Elétrons e prótons são eletricamente carregados: 1,60 x 10-19 C; Elétrons:
Física Moderna II - FNC376
 Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto 1o. Semestre de 2008 1 Tabela Periódica dos elementos 2 8 8 Alcalinos, um elétron livre na camada
Universidade de São Paulo Instituto de Física Física Moderna II - FNC376 Profa. Márcia de Almeida Rizzutto 1o. Semestre de 2008 1 Tabela Periódica dos elementos 2 8 8 Alcalinos, um elétron livre na camada
A teoria de Hartree. r r. r r
 A teoria de Hartree Vimos, na aula 4, a proposta de Hartree: tratamento autoconsistente Ou seja, V(r) obtido a posteriori, a partir das distribuições de carga dos e -, deve concordar com o V(r) usado para
A teoria de Hartree Vimos, na aula 4, a proposta de Hartree: tratamento autoconsistente Ou seja, V(r) obtido a posteriori, a partir das distribuições de carga dos e -, deve concordar com o V(r) usado para
Fundamentos da modelagem Molecular - 1
 Fundamentos da modelagem Molecular - 1 Departamento de Física UFPel ufpellogo Introdução Rotas para a pesquisa Conexão entre experimento, simulação e teoria Sistema real Fazer modelos do sistema Fazer
Fundamentos da modelagem Molecular - 1 Departamento de Física UFPel ufpellogo Introdução Rotas para a pesquisa Conexão entre experimento, simulação e teoria Sistema real Fazer modelos do sistema Fazer
Teoria dos Orbitais Moleculares. Prof. Fernando R. Xavier
 Teoria dos Orbitais Moleculares Prof. Fernando R. Xavier UDESC 013 Antecedentes... A teoria de ligação de valência (TLV) não cosnsegue explicar com eficiência a formação de moléculas poliatômicas. Uma
Teoria dos Orbitais Moleculares Prof. Fernando R. Xavier UDESC 013 Antecedentes... A teoria de ligação de valência (TLV) não cosnsegue explicar com eficiência a formação de moléculas poliatômicas. Uma
Estrutura Atômica I. Química Quântica Prof a. Dr a. Carla Dalmolin. Átomo de Hidrogênio Átomos Hidrogenóides
 Estrutura Atômica I Química Quântica Prof a. Dr a. Carla Dalmolin Átomo de Hidrogênio Átomos Hidrogenóides Aplicações da Mecânica Quântica Soluções da Equação de Schrödinger independente do tempo Partícula
Estrutura Atômica I Química Quântica Prof a. Dr a. Carla Dalmolin Átomo de Hidrogênio Átomos Hidrogenóides Aplicações da Mecânica Quântica Soluções da Equação de Schrödinger independente do tempo Partícula
Aula 01 Estrutura eletrônica
 Universidade Tecnológica Federal do Paraná Departamento Acadêmico de Química e Biologia Aula 01 Estrutura eletrônica Dr. Tiago P. Camargo Átomos - Estrutura Núcleo (prótons e nêutrons) Eletrosfera (elétrons)
Universidade Tecnológica Federal do Paraná Departamento Acadêmico de Química e Biologia Aula 01 Estrutura eletrônica Dr. Tiago P. Camargo Átomos - Estrutura Núcleo (prótons e nêutrons) Eletrosfera (elétrons)
Física IV Poli Engenharia Elétrica: 20ª Aula (04/11/2014)
") Física IV Poli Engenharia Elétrica: ª Aula (4/11/14) Prof. Alvaro Vannucci a última aula vimos: Átomos multi-eletrônicos: as energias dos estados quânticos podem ser avaliadas através da expressão: 13,6
Física IV Poli Engenharia Elétrica: ª Aula (4/11/14) Prof. Alvaro Vannucci a última aula vimos: Átomos multi-eletrônicos: as energias dos estados quânticos podem ser avaliadas através da expressão: 13,6
FF-296: Teoria do Funcional da Densidade I. Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785
 FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Teorema de Koopmans Gap e descontinuidade na derivada Teorema de
FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Teorema de Koopmans Gap e descontinuidade na derivada Teorema de
Fundamentos de Química Quântica
 Universidade Federal de Ouro Preto Instituto de Ciências Exatas e Biológicas Departamento de Química Fundamentos de Química Quântica Aula 3 Professora: Melissa Soares Caetano Átomo de Hidrogênio Um núcleo
Universidade Federal de Ouro Preto Instituto de Ciências Exatas e Biológicas Departamento de Química Fundamentos de Química Quântica Aula 3 Professora: Melissa Soares Caetano Átomo de Hidrogênio Um núcleo
n Camadas K L M N...
 Notação Espectroscópica para Configurações Eletrônicas em Átomos. Modelo de Partícula Independente. Podemos obter uma quantidade expressiva importante de informações sobre estados atômicos de sistemas
Notação Espectroscópica para Configurações Eletrônicas em Átomos. Modelo de Partícula Independente. Podemos obter uma quantidade expressiva importante de informações sobre estados atômicos de sistemas
O Modelo de Bose-Hubbard
 O Modelo de Bose-Hubbard Alvaro Montaña Guerrero Teoría quântica de muitos corpos Instituto de Física Universidade de São Paulo SP, Brasil 10 de Dezembro de 2015 O Modelo de Bose-Hubbard 1 / 21 Introdução
O Modelo de Bose-Hubbard Alvaro Montaña Guerrero Teoría quântica de muitos corpos Instituto de Física Universidade de São Paulo SP, Brasil 10 de Dezembro de 2015 O Modelo de Bose-Hubbard 1 / 21 Introdução
A teoria dos orbitais moleculares
 A teoria dos orbitais moleculares Qualitativamente, indica regiões do espaço (entre os átomos que formam uma molécula) nas quais a probabilidade de encontrar os elétrons é máxima. Na mecânica quântica,
A teoria dos orbitais moleculares Qualitativamente, indica regiões do espaço (entre os átomos que formam uma molécula) nas quais a probabilidade de encontrar os elétrons é máxima. Na mecânica quântica,
Química Orgânica Ambiental
 Química Orgânica Ambiental Aula 1 Estrutura Eletrônica e ligação química Prof. Dr. Leandro Vinícius Alves Gurgel 1. Introdução: O átomo Os átomos são formados por nêutrons, prótons e elétrons: Prótons
Química Orgânica Ambiental Aula 1 Estrutura Eletrônica e ligação química Prof. Dr. Leandro Vinícius Alves Gurgel 1. Introdução: O átomo Os átomos são formados por nêutrons, prótons e elétrons: Prótons
Interações Atômicas e Moleculares
 Interações Atômicas e Moleculares 6. O Princípio Variacional Prof. Pieter Westera pieter.westera@ufabc.edu.br http://professor.ufabc.edu.br/~pieter.westera/iam.html Na aula anterior, tratando de moléculas
Interações Atômicas e Moleculares 6. O Princípio Variacional Prof. Pieter Westera pieter.westera@ufabc.edu.br http://professor.ufabc.edu.br/~pieter.westera/iam.html Na aula anterior, tratando de moléculas
Estrutura dos Materiais. e Engenharia dos Materiais Prof. Douglas Gouvêa
 Ligações Químicas e Estrutura dos Materiais PMT 5783 - Fundamentos de Ciência e Engenharia dos Materiais Prof. Douglas Gouvêa Objetivos Descrever a estrutura atômica e suas conseqüências no tipo de ligação
Ligações Químicas e Estrutura dos Materiais PMT 5783 - Fundamentos de Ciência e Engenharia dos Materiais Prof. Douglas Gouvêa Objetivos Descrever a estrutura atômica e suas conseqüências no tipo de ligação
Átomos multi-eletrônicos. Indistinguibilidade
 Átomos multi-eletrônicos Indistinguibilidade Princípio de exclusão, de Pauli 1. Em um átomo multi-eletrônico nunca pode haver mais de 1 e- ocupando o mesmo estado quântico.. Um sistema constituído de vários
Átomos multi-eletrônicos Indistinguibilidade Princípio de exclusão, de Pauli 1. Em um átomo multi-eletrônico nunca pode haver mais de 1 e- ocupando o mesmo estado quântico.. Um sistema constituído de vários
Aula 12. (quase) Tudo sobre os átomos. Física Geral F-428
 Tudo sobre os átomos. Física Geral F-428") Aula 1 (quase) Tudo sobre os átomos Física Geral F-48 1 Algumas propriedades atômicas: Átomos são estáveis (quase sempre); Os átomos podem ser agrupados em famílias (propriedades periódicas, com o número
Aula 1 (quase) Tudo sobre os átomos Física Geral F-48 1 Algumas propriedades atômicas: Átomos são estáveis (quase sempre); Os átomos podem ser agrupados em famílias (propriedades periódicas, com o número
QUÍMICA A Ciência Central 9ª Edição Capítulo 8 Conceitos básicos de ligação química David P. White
 QUÍMICA A Ciência Central 9ª Edição Capítulo 8 Conceitos básicos de ligação química David P. White Ligações químicas, símbolos de Lewis e a regra do octeto Ligação química: é a força atrativa que mantém
QUÍMICA A Ciência Central 9ª Edição Capítulo 8 Conceitos básicos de ligação química David P. White Ligações químicas, símbolos de Lewis e a regra do octeto Ligação química: é a força atrativa que mantém
Introdução à Modelagem Molecular
 Introdução à Modelagem Molecular Prof. Alex Fabiano C. Campos, Dr Introdução Métodos teóricos e técnicas computacionais para modelar ou imitar o comportamento de moléculas e sistemas atômicos. Empregados
Introdução à Modelagem Molecular Prof. Alex Fabiano C. Campos, Dr Introdução Métodos teóricos e técnicas computacionais para modelar ou imitar o comportamento de moléculas e sistemas atômicos. Empregados
Quantização. Quantização da energia (Planck, 1900) hc h. Efeito fotoelétrico (Einstein, 1905) Espectros atômicos (linhas discretas) v 2
 hc h. Efeito fotoelétrico (Einstein, 1905) Espectros atômicos (linhas discretas) v 2") Mecânica Quântica Quantização e o modelo de Bohr (revisão) Dualidade Onda-Partícula Princípio da Incerteza Equação de Schrödinger Partícula na Caixa Átomo de Hidrogênio Orbitais Atômicos Números Quânticos
Mecânica Quântica Quantização e o modelo de Bohr (revisão) Dualidade Onda-Partícula Princípio da Incerteza Equação de Schrödinger Partícula na Caixa Átomo de Hidrogênio Orbitais Atômicos Números Quânticos
Eletromagnetismo Aplicado
 Eletromagnetismo Aplicado Unidade 3 Prof. Marcos V. T. Heckler 1 Conteúdo Introdução Materiais dielétricos, polarização e permissividade elétrica Materiais magnéticos, magnetização e permeabilidade magnética
Eletromagnetismo Aplicado Unidade 3 Prof. Marcos V. T. Heckler 1 Conteúdo Introdução Materiais dielétricos, polarização e permissividade elétrica Materiais magnéticos, magnetização e permeabilidade magnética
Física Molecular Estrutura das Ligações Químicas
 Física Molecular Estrutura das Ligações Químicas 100 É razoável focalizar o estudo de moléculas nas interações entre elétrons e núcleos já reagrupados em caroço atômico (núcleo + camadas fechadas internas),
Física Molecular Estrutura das Ligações Químicas 100 É razoável focalizar o estudo de moléculas nas interações entre elétrons e núcleos já reagrupados em caroço atômico (núcleo + camadas fechadas internas),
Aula anterior. Equação de Schrödinger a 3 dimensões. d x 2m - E -U. 2m - E -U x, y, z. x y z x py pz cin cin. E E ( x, y,z ) - 2m 2m x y z
 - 2m 2m x y z") 6/Maio/2013 Aula 21 Efeito de túnel quântico: decaimento alfa. Aplicações: nanotecnologias; microscópio por efeito de túnel. Equação de Schrödinger a 3 dimensões. Átomo de hidrogénio Modelo de Bohr 8/Maio/2013
6/Maio/2013 Aula 21 Efeito de túnel quântico: decaimento alfa. Aplicações: nanotecnologias; microscópio por efeito de túnel. Equação de Schrödinger a 3 dimensões. Átomo de hidrogénio Modelo de Bohr 8/Maio/2013
1.1. Motivação Objetivos
 1 Introdução As Unidades de Processamento Gráfico (GPUs), ou Placas Gráficas, foram originalmente desenvolvidas com o propósito de renderização gráfica. Contudo, nos últimos anos, o desempenho na realização
1 Introdução As Unidades de Processamento Gráfico (GPUs), ou Placas Gráficas, foram originalmente desenvolvidas com o propósito de renderização gráfica. Contudo, nos últimos anos, o desempenho na realização
Sabrina Silva Carara
 Estudo de Defeitos em Camadas de Grafeno usando Dinâmica Molecular por Primeiros Princípios Sabrina Silva Carara Sabrina Silva Carara Estudo de Defeitos em Camadas de Grafeno usando Dinâmica Molecular
Estudo de Defeitos em Camadas de Grafeno usando Dinâmica Molecular por Primeiros Princípios Sabrina Silva Carara Sabrina Silva Carara Estudo de Defeitos em Camadas de Grafeno usando Dinâmica Molecular
ESCOLA SECUNDÁRIA PINHAL DO REI FICHA FORMATIVA 2ºTESTE
 Ano Letivo 2016/2017 ESCOLA SECUNDÁRIA PINHAL DO REI FICHA FORMATIVA 2ºTESTE Física e Química A 10ºAno 1. A energia dos eletrões nos átomos inclui: (A) apenas o efeito das atrações entre os eletrões e
Ano Letivo 2016/2017 ESCOLA SECUNDÁRIA PINHAL DO REI FICHA FORMATIVA 2ºTESTE Física e Química A 10ºAno 1. A energia dos eletrões nos átomos inclui: (A) apenas o efeito das atrações entre os eletrões e
Tabela Periódica dos elementos
 Tabela Periódica dos elementos 8 8 Alcalinos, um elétron livre na camada (pode ser facilmente removido), bons condutores Todos possuem subcamada fechada 18 18 3 1 ATENÇÃO 0 Ca (Z=0 A=40) tem camadas completas,
Tabela Periódica dos elementos 8 8 Alcalinos, um elétron livre na camada (pode ser facilmente removido), bons condutores Todos possuem subcamada fechada 18 18 3 1 ATENÇÃO 0 Ca (Z=0 A=40) tem camadas completas,
QUI109 QUÍMICA GERAL (Ciências Biológicas) 7ª aula /
 7ª aula /") QUI109 QUÍMICA GERAL (Ciências Biológicas) 7ª aula / 2016-2 Prof. Mauricio X. Coutrim (disponível em: http://professor.ufop.br/mcoutrim) LIGAÇÃO QUÍMICA É A FORÇA QUE MANTÉM ÁTOMOS E/OU ÍONS UNIDOS NAS
QUI109 QUÍMICA GERAL (Ciências Biológicas) 7ª aula / 2016-2 Prof. Mauricio X. Coutrim (disponível em: http://professor.ufop.br/mcoutrim) LIGAÇÃO QUÍMICA É A FORÇA QUE MANTÉM ÁTOMOS E/OU ÍONS UNIDOS NAS
FF-296: Teoria do Funcional da Densidade I. Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785
 FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tópicos Escala Funções de onda Funcionais da densidade Termo de
FF-296: Teoria do Funcional da Densidade I Prof. Dr. Ronaldo Rodrigues Pelá Sala 2602A-1 Ramal 5785 rrpela@ita.br www.ief.ita.br/~rrpela Tópicos Escala Funções de onda Funcionais da densidade Termo de
Gases reais. ρ(γ) = ρ(q 1,q 2,...,q 3N,p 1,p 2,...,p 3N ) (1)
 = ρ(q 1,q 2,...,q 3N,p 1,p 2,...,p 3N ) (1)") Capítulo 7 Gases reais 1 Distribuição canônica Diferentemente do que foi feito nos capítulos anteriores vamos considerar agora sistemas de partículas interagentes. Por exemplo, um gás composto de N moléculas
Capítulo 7 Gases reais 1 Distribuição canônica Diferentemente do que foi feito nos capítulos anteriores vamos considerar agora sistemas de partículas interagentes. Por exemplo, um gás composto de N moléculas
Descoberta do Núcleo
 Unidade 3 Núcleo Atômico Descoberta do Núcleo Propriedades dos Núcleos Forças Nucleares Estabilidade Nuclear Ressonância Magnética Nuclear Consultas http://hyperphysics.phy-astr.gsu.edu/hbase/nuccon.html#nuccon
Unidade 3 Núcleo Atômico Descoberta do Núcleo Propriedades dos Núcleos Forças Nucleares Estabilidade Nuclear Ressonância Magnética Nuclear Consultas http://hyperphysics.phy-astr.gsu.edu/hbase/nuccon.html#nuccon
Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais
 Ligações Químicas Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais Ligação iônica Transferência de elétrons de um átomo para outro Íons de cargas opostas Forças
Ligações Químicas Ligações Interatômicas: IÔNICA = metal + não-metal COVALENTE = não-metais METÁLICA = metais Ligação iônica Transferência de elétrons de um átomo para outro Íons de cargas opostas Forças
Estrutura molecular Ligação química
 Estrutura molecular Ligação química A grande diversidade de materiais que nos rodeia tem origem na variedade de substâncias que os constituem. Esta variedade e diversidade resulta das diferentes combinações
Estrutura molecular Ligação química A grande diversidade de materiais que nos rodeia tem origem na variedade de substâncias que os constituem. Esta variedade e diversidade resulta das diferentes combinações
A energia de formação para o B em um sítio de Si ou C no bulk é dada pelas seguintes
 63 Figura 22: Estrutura de bandas. Em (a) o 3C-SiC bulk puro, (b) com um átomo de B substituindo um átomo de Si e em (c) B ocupando a posição de um átomo de C. O topo da banda de valência foi tomado como
63 Figura 22: Estrutura de bandas. Em (a) o 3C-SiC bulk puro, (b) com um átomo de B substituindo um átomo de Si e em (c) B ocupando a posição de um átomo de C. O topo da banda de valência foi tomado como
Física Moderna II Aula 08
 Universidade de São Paulo Instituto de Física 1 º Semestre de 2015 Profa. Márcia de Almeida Rizzutto Oscar Sala sala 220 rizzutto@if.usp.br Física Moderna II Aula 08 Monitor: Gabriel M. de Souza Santos
Universidade de São Paulo Instituto de Física 1 º Semestre de 2015 Profa. Márcia de Almeida Rizzutto Oscar Sala sala 220 rizzutto@if.usp.br Física Moderna II Aula 08 Monitor: Gabriel M. de Souza Santos
Disciplina: Química Professor: Rubens Barreto. III Unidade
 Disciplina: Química Professor: Rubens Barreto III Unidade Ligações Químicas Ligações iônicas Tipos de Ligações Ligações covalentes Ligações metálicas Os gases nobres e a regra do octeto Todas as substâncias
Disciplina: Química Professor: Rubens Barreto III Unidade Ligações Químicas Ligações iônicas Tipos de Ligações Ligações covalentes Ligações metálicas Os gases nobres e a regra do octeto Todas as substâncias
A Teoria de Orbitais Moleculares
 2 A Teoria de Orbitais Moleculares 2.1 A Equação de Schroedinger De acordo com a mecânica clássica de Hamilton, um sistema de partículas interagentes tem energia total : T + V = E A mecânica quântica de
2 A Teoria de Orbitais Moleculares 2.1 A Equação de Schroedinger De acordo com a mecânica clássica de Hamilton, um sistema de partículas interagentes tem energia total : T + V = E A mecânica quântica de
TEORIA DOS ORBITAIS MOLECULARS. QFL QUIMICA GERAL 1 (1o sem 2017)
") TEORIA DOS ORBITAIS MOLECULARS QFL1101 - QUIMICA GERAL 1 (1o sem 017) Teoria de Ligação pela Valência Linus Pauling; Os elétrons de valência estão localizados entre os átomos (ou são pares isolados); Orbitais
TEORIA DOS ORBITAIS MOLECULARS QFL1101 - QUIMICA GERAL 1 (1o sem 017) Teoria de Ligação pela Valência Linus Pauling; Os elétrons de valência estão localizados entre os átomos (ou são pares isolados); Orbitais
Química A Extensivo V. 8
 Química A Extensivo V. Exercícios 0) A α 90 Th3 Ra 9 Ac 90 Th α Ra Número de massa: Prótons: Neutons: 36 0) 90 Th 3 α Ra α 6 Rn α Po 0 5 At 0 03) B 0) C Prótons: 5 Elétons: 5 Neutons: 00 5 = 35 6X O elemento
Química A Extensivo V. Exercícios 0) A α 90 Th3 Ra 9 Ac 90 Th α Ra Número de massa: Prótons: Neutons: 36 0) 90 Th 3 α Ra α 6 Rn α Po 0 5 At 0 03) B 0) C Prótons: 5 Elétons: 5 Neutons: 00 5 = 35 6X O elemento
Universidade de São Paulo Instituto de Física. Física Moderna II. Profa. Márcia de Almeida Rizzutto 2 o Semestre de Física Moderna 2 Aula 8
 Universidade de São Paulo Instituto de Física Física Moderna II Profa. Márcia de Almeida Rizzutto o Semestre de 04 Átomos multi-eletrônicos Indistinguibilidade Princípio de exclusão, de Pauli. Em um átomo
Universidade de São Paulo Instituto de Física Física Moderna II Profa. Márcia de Almeida Rizzutto o Semestre de 04 Átomos multi-eletrônicos Indistinguibilidade Princípio de exclusão, de Pauli. Em um átomo
Ligações químicas e VSEPR
 BIK0102: ESTRUTURA DA MATÉRIA Crédito: Sprace Ligações químicas e VSEPR Professor Hugo Barbosa Suffredini hugo.suffredini@ufabc.edu.br Site: www.suffredini.com.br Ligações Químicas IÔNICA CVALENTE METÁLICA
BIK0102: ESTRUTURA DA MATÉRIA Crédito: Sprace Ligações químicas e VSEPR Professor Hugo Barbosa Suffredini hugo.suffredini@ufabc.edu.br Site: www.suffredini.com.br Ligações Químicas IÔNICA CVALENTE METÁLICA