ESTUDO GENÉTICO-CLÍNICO DE DEFICIENTES MENTAIS INSTITUCIONALIZADOS.
|
|
|
- Luiz Gustavo Sampaio Cunha
- 6 Há anos
- Visualizações:
Transcrição
1 UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO ESTUDO GENÉTICO-CLÍNICO DE DEFICIENTES MENTAIS INSTITUCIONALIZADOS. Lisandra Mesquita Batista Dissertação de Mestrado apresentada ao Departamento de Genética da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, para obtenção do grau de Mestre em Genética. Orientador: Prof. Dr. João Monteiro de Pina Neto
2 Livros Grátis Milhares de livros grátis para download.
3 UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO Estudo genético-clínico de deficientes mentais institucionalizados Lisandra Mesquita Batista RIBEIRÃO PRETO 2008
4 UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO Estudo genético-clínico de deficientes mentais institucionalizado Dissertação de Mestrado apresentada ao Departamento de Genética da faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, para obtenção do grau de Mestre em Genética. Lisandra Mesquita Batista Orientador: Prof. Dr. João Monteiro de Pina Neto RIBEIRÃO PRETO 2008
5 FICHA CATALOGRÁFICA Batista, Lisandra Mesquita Estudo genético-clínico de pacientes institucionalizados FMRP-USP. Ribeirão Preto, p,: il.; 30cm Tese (Mestrado) Faculdade de Medicina de Ribeirão Preto - Universidade de São Paulo Departamento de Genética. Orientador: Prof. Dr. João Monteiro de Pina-Neto. 1. Deficiência Mental. 2. Diagnóstico clínico. 3.Diagnóstico etiológico.
6 Aos meus pais, Lélio e Rosângela Aos meus irmãos, Lélio, Lorena e Leonardo
7 AGRADECIMENTOS Agradeço a todos que ao meu lado estiveram com apoio e aprovação contribuindo para minha formação profissional. Ao Prof. Dr. João Monteiro de Pina Neto pelos comentários inteligentes, conselhos, atenção, paciência e por nunca permitir que eu perdesse o foco dos meus objetivos. Agradeço, ainda, por cada palavra de incentivo e por perpetuar minha admiração pela genética clínica. À Liane de Rosso Giuliani pela cumplicidade neste trabalho com APAEs. Às APAEs de Altinópolis e Serrana que possibilitaram este estudo e acreditaram na importância da genética. A todos os profissionais dessas APAEs pela amizade e dedicação. Aos colegas do Serviço de Genética Médica do Hospital das Clínicas de Ribeirão Preto, sempre dispostos a discutir casos e auxiliar nas solicitações de exames necessários para o estudo desses pacientes. Aos profissionais do Laboratório de Citogenética Humana e Genética Molecular do Prof. Dr. João Monteiro de Pina-Neto pela parceria e cooperação. Aos funcionários do Departamento de Genética da Faculdade de Medicina de Ribeirão Preto pela atenção.
8 Aos docentes de outros serviços, principalmente do Departamento de Radiologia do HCFMRP-USP que disponibilizaram do seu tempo para discussão de casos e exames. Ao Rafael Jannarelli Ulson pelo otimismo contagiante e companherismo.
9 SUMÁRIO RESUMO ABSTRACT 1. INTRODUÇÃO JUSTIFICATIVA OBJETIVOS Objetivo geral Objetivo específico CASUÍSTICA E MÉTODOS Classificação Clínica Classificação Etiológica RESULTADOS Geral Classificação quanto ao nível de DM Classificações Clínica e Etiológica das DMs Classificação Etiológica e nível de DM Exames Complementares Citogenética...45
10 Exame de Imagem do Sistema Nervoso Central Exame para Erros Inatos do Metabolismo Exame Molecular DISCUSSÃO Geral Classificação quanto ao nível de DM Classificação Clínica e Etiológica Classificação Etiológica e nível de DM Caracterização dos Exames Complementares CONCLUSÕES...71 ANEXO...73 BIBLIOGRAFIA...82
11 RESUMO A Deficiência Mental (DM) é uma preocupação para a saúde pública e a sociedade, sendo a pesquisa de sua etiologia um desafio para a medicina. Estima-se que 10% da população em países em desenvolvimento, são portadores de algum tipo de deficiência, sendo que metade destes são portadores de deficiência mental. Com o objetivo de se estabelecer o diagnóstico clínico e etiológico da DM em uma população de deficientes mentais institucionalizados, foram avaliados 200 pacientes das APAEs de Altinópolis e Serrana. Para isso, foi realizado um exame genético-clínico com construção dos heredogramas e exames complementares, como o de imagem do Sistema Nervoso Central (SNC), exames metabólicos, cariótipo e análise molecular para as síndromes do FRAXA e FRAXE. Esses exames complementares mostraram-se relevantes e com valor diagnóstico. Foram selecionados para exame de cariótipo 91 pacientes, entre eles as síndromes reconhecidas, as prováveis síndromes e os casos indefinidos, sendo observado 30% de alterações; para triagem molecular FRAXA e FRAXE foram selecionados os casos do sexo masculino sem causa definida, o que correspondeu a 35 exames, observando-se 1 (2,8%) caso positivo; para triagem de erros inatos do metabolismo, 17 casos suspeitos foram selecionados e foi notado alteração em 35%; para imagem do SNC, os casos com sintomas neurológicos, alterações cranianas e casos sem definição de DM foram selecionados e observou-se 69% de anormalidades. Os pacientes foram classificados quanto a seu diagnóstico, clinicamente e etiologicamente. Classificou-se, clinicamente, 23% dos pacientes com DM com anomalias múltiplas, 4% DM com malformação do SNC isolada, 3% DM e erros inatos do metabolismo, 42% com DM de causa ambiental, 1% DM e disfunção do SNC, 7% DM aparentemente puro e 20% DM não classificado. Quanto a etiologia, 28% dos
12 pacientes apresentaram DM de causa genética (14% anomalias cromossômicas e 14% anomalias gênicas), 42% de causa ambiental, 4% heterogêneo, 2% esporádico, 1% multifatorial e 23% indefinido. O perfil desta amostra evidenciou uma prevalência do sexo masculino com uma razão de 1.3 M:1 F. Observou-se consangüinidade em 1,5% dos casos. Houve predomínio de DM moderada (48,5%) e grave (22,5%). Estabelecer o diagnóstico etiológico de DM é complexo e continua sendo um desafio para os diferentes especialistas, mas mostra-se essencial principalmente para definir estratégias de prevenção e manejo das deficiências mentais.
13 ABSTRACT Mental Retardation (MR) is a key concern for public health and society, and the research about its etiology is a challenge for Medicine. It is estimated that 10 % of the population in developing countries are of disabled people, half of which are mentally disabled. Aiming at establishing the clinical and etiological diagnostic of MD in a population of institutionalized mental deficients, 200 patients of the APAE institution in the cities of Altinópolis and Serrana were evaluated. For that, it was carried out a geneticoclinical exam with formation of kin relationships (heredograms) and complementary exams, such as the Central Nervous System (CN), metabolic exams, cariotype and molecular analysis for the FRAXA and FRAXE syndromes. These complementary exams demonstrated to be relevant and to have diagnostic value. It has been selected 91 patients for the cariotype exam having either recognized syndromes, probable syndromes or undefined cases, with 30 % of alterations observed. For the FRAXA and FRAXE molecular screening, it has been selected the male-sex cases without a defined cause, corresponding to 35 exams of which only one positive case (2.8 %) was observed. For the screening of metabolism inborn errors, 17 suspected cases were selected and it has been noted alteration in 35 % of them. For the CNS screening, the cases with neurological symptoms, cranial alterations and MR undefined cases were selected and it has been observed 69 % of abnormalities. The patients were classified with respect to their clinical and etiological diagnosis. Twenty three percent of the patients were classified clinically with multiple anomalies MR, 4 % with isolated-cns malformation MR, 3 % with MR and metabolism inborn errors, 42 % with MR from environmental causes, 1 % with MR and CNS dysfunction, 7 % with apparently pure MR and 20 % with unclassified MR. Concerning etiology, 28 % of the patients presented genetic MR (14 % cromossomic anomalies and 14 % genetic
14 anomalies), 42 % environmental cause, 4 % heterogeneous, 2 % sporadic, 1 % multifactorial and 23 % undefined. The profile of such sample evidenced a prevalence of the male (M) sex over the female (F) sex with a ratio of 1.3:1 (M:F). It has been observed consanguinity in 1.5 % of the cases. There has been prevalence of moderate MR (48.5 %) and grave MR (22.5 %). Establishing the etiological diagnosis of MR is complex and continues to be a challenge for different specialists; nevertheless, it is essential mainly in defining strategies for preventing and handling with mental deficiencies.
15 1 INTRODUÇÃO A Deficiência Mental (DM) é uma condição de grande preocupação para a saúde pública e a sociedade, sendo a pesquisa de sua etiologia um desafio para a medicina. A DM envolve uma série de problemas médicos-biológicos e psico-sociais complexos, entretanto, tem havido um grande progresso na elucidação da sua etiologia. Os estudos sobre as Deficiências Mentais iniciaram-se no começo do século XX com a publicação do primeiro teste de avaliação da função mental de Binet e Simon em Hoje, está em discussão este termo Deficiência Mental, pois como sugere Warren (2002), tem promovido estigmas e estereotipias. Ao longo da história, muitos conceitos existiram e a pessoa com esta deficiência já foi chamada, nos círculos acadêmicos, por vários nomes: oligofrênica, débil profunda, criança excepcional, criança subnormal, mongolóide, criança atrasada, entre outros. Mas, atualmente, quanto ao nome da condição, há uma tendência mundial de se usar o termo Deficiência Intelectual (Sassaki, 2005). Este termo seria mais apropriado por referir-se ao funcionamento do intelecto especificamente e não ao funcionamento da mente como um todo e também por melhor distinguir entre deficiência mental e doença mental, dois termos que geram confusão há vários séculos. Embora esta seja uma tendência, encontra-se o termo Deficiência Mental com maior freqüência. A primeira definição de Deficiência Mental foi apresentada por Tredgold em 1908 que enfatizava ser uma condição incurável e de status permanente. Estabelecia que a DM era um estado de defeito mental a partir do nascimento ou idade mais precoce em função do desenvolvimento cerebral incompleto, e em conseqüência disso, a pessoa afetada se tornava incapaz de desempenhar suas tarefas como membro da sociedade. Houveram várias atualizações
16 das definições de DM, mas todas tendo como base apenas o score do Quociente de Inteligência QI, o que mostrou-se insuficiente e passível de erro. Em 1992, foi publicado pela AAMR (Associação Americana de Retardo Mental) uma definição de natureza mais funcional, levandose em conta o comportamento adaptativo. Portanto, o conceito atual de deficiência mental (DM) vê cada indivíduo de forma global e funcional, o que significa transpor o conjunto de condições apresentado por ele para a sua interação com o ambiente. Foca-se na capacidade de adaptação da pessoa ao mundo, que é o elemento mais fortemente relacionado à noção de normal. Segundo a AAMR e DSM-IV (Manual Diagnóstico e Estatístico de Transtornos Mentais), por deficiência mental entende-se o estado de redução notável do funcionamento intelectual significativamente inferior à média, associado a limitações em pelo menos dois aspectos do funcionamento adaptativo: comunicação, cuidados pessoais, competências domésticas, habilidades sociais, utilização dos recursos comunitários, autonomia, saúde e segurança, aptidões escolares, lazer e trabalho, que se manifesta antes dos 18 anos de idade. Segundo a Organização Mundial de Saúde, 10% da população em países em desenvolvimento, são portadores de algum tipo de deficiência, sendo que metade destes são portadores de deficiência mental. Em países desenvolvidos ocorre em 2 a 3 % da população geral e considerando a definição de deficiência mental pelo DSM-IV, relata-se freqüência de 3 % para DM leve e 0,4 % para DM grave. No Brasil não há dados estatísticos confiáveis nesta área, mas há relatos de que as deficiências atingiriam até 10% da população geral (Krinsky, 1983). Há predominância de homens com DM em relação às mulheres, independentemente do grau de DM, do país de estudo ou das condições sócio-econômicas. Hoje se considera uma razão média de prevalência homem : mulher de 1,3 : 1 (Frints et al., 2002). Essa predominância pode ser explicada devido à presença de genes no cromossomo X relacionados com a estrutura e função
17 cerebral (Lehrke et al., 1974; Morton et al., 1992; Turner et al., 1996; Lubs et al., 1999; Plomin et al., 2004; Skuse et al., 2005), e pelo fato das diferenças biológicas entre homens e mulheres, conferidas pelo número diferente de cromossomos sexuais. A classificação da DM pode ser abordada de várias formas: - nível mental (proposta pela OMS) Coeficiente Denominação Nível cognitivo Idade mental intelectual segundo Piaget correspondente Menor de 20 Profundo Período Sensório-Motriz Entre 20 e 35 Grave Período Sensório-Motriz Entre 36 e 51 Moderado Período Préoperativo Entre 52 e 67 Leve Período das Operações Concretas 0-2 anos 0-2 anos 2-7 anos 7-12 anos Este sistema quantitativo (de QI ) é muito questionado. Baseado na definição atual de DM aconselha-se um método qualitativo, dando privilégio aos critérios adaptativos, mais que nos índices numéricos de QI, para se especificar o grau de comprometimento funcional. Importa mais saber se a pessoa com Deficiência Mental necessita de apoio em habilidades de comunicação, em habilidades sociais, etc, mais que em outras áreas. Esse novo enfoque centraliza-se mais no
18 indivíduo deficiente, independentemente de seu escore de QI, sob o ponto de vista das oportunidades e autonomias. -etiológica Compreende as diferentes etiologias: genética, ambiental, multifatorial e desconhecida. Existe uma grande variedade etiológica, e também, a permanência de um grande número de casos (20 a 50 %) sem etiologia definida (Van Buggenhout et al., 2001). Opitz propôs, em 1977, grupos etiopatogênicos que possibilitaram uma divisão mais específica da deficiência mental. GRUPO DM com malformações congênitas múltiplas DM com malformação isolada do SNC DM de causa ambiental DM e erros inatos do metabolismo DM e psicoses DM e disfunção do SNC (epilepsia, paralisia cerebral, hipotonia) DM aparentemente puro DM não classificado O diagnóstico de deficiência mental é muitas vezes difícil. Essa grande diversidade de etiologias e processos patológicos em tantos quadros diferentes, pode dificultar o trabalho do médico que faz a avaliação inicial do paciente com DM, principalmente nos casos mais graves (Battaglia e Carey, 2003). Numerosos fatores emocionais, alterações de certas atividades nervosas superiores, como atraso específico de linguagem ou dislexia, psicoses ou baixo nível sócio econômico ou cultural podem estar na base da impossibilidade do ajustamento social adaptativo adequado, sem que haja necessariamente deficiência mental. Estes fatores devem ser
19 levados em conta e, portanto, adequadamente diagnosticados quando uma criança suspeita de ter uma deficiência mental é submetida à avaliação de sua capacidade intelectual permitindo a avaliação das possibilidades de inserção social da criança e orientando a abordagem terapêutica e educacional. Recentemente, o consenso da conferência do American College of Medical Genetics estabeleceu diretrizes em relação à avaliação de pacientes com DM, enfatizando a grande importância dos exames de citogenética e neuroimagem no diagnóstico desses pacientes, mas, principalmente, ressaltando a necessidade de uma anamnese adequada e do exame físico completo (Curry et al., 1997). A deficiência mental é uma condição complexa, sendo de grande importância que seu diagnóstico seja realizado, pois destina-se a finalidades diversas, como elegibilidade para intervenção; benefícios e assistência previdenciária; proteção legal; acesso a cotas para emprego e outras. Conjuntamente, há necessidade de se estabelecer o diagnóstico clínico e etiológico da deficiência mental para melhor orientação do tratamento, da habilitação e, principalmente, da prevenção primária. Desse modo, como instrumento clínico e legal, o diagnóstico deve estar incorporado às práticas do atendimento nas deficiências.
20 Segue abaixo um fluxograma que propõe uma investigação de deficiência mental: INVESTIGAÇÃO DIAGNÓSTICA DE DEFICIÊNCIA MENTAL ADNPM / DM Avaliação Clínica Completa Neuroimagem Sorologias TORCH Audiometria Fundoscopia Suspeita de quadro sindrômico de etiologia monogênica, incluindo EIM DM com dismorfias maiores e/ou menores sem especificações DM isolado Investigação específica cariótipo Pesquisa FRAXA Pesquisa de perdas subteloméricas + - alterado normal + - normal DM de etiologia definida DM de etiologia indeterminada Levando-se em conta as melhorias no atendimento ao parto, o controle de doenças transmissíveis e da subnutrição de gestantes e recém-nascidos, assim como nas condições gerais de vida, a doença genética tornou-se uma questão prioritária de saúde pública. Além disso, a sobrevida dos pacientes com enfermidades genéticas está aumentando, pressionando a sociedade no sentido de maior custo na área da saúde. Estima-se que o custo médio de vida de uma pessoa
21 com DM seja de dólares americanos, segundo o Centro de Prevenção e Controle de Doenças dos Estados Unidos (2003), sendo 14% gastos com saúde (atendimento médico, hospitais, medicações); 10% com modificações da casa e educação especial; 76%gastos indiretos (limitação como mão-de-obra para o país). Dissemina-se, hoje, que as APAEs desenvolvam ações voltadas para os três níveis de prevenção. Na prevenção primária, desenvolve-se ações para evitar-se que uma criança deficiente seja gerada, como ocorre com o aconselhamento genético. Este pode ser definido como um processo de comunicação sobre o risco de ocorrência ou recorrência familial de doenças genéticas, com as finalidades de fornecer a indivíduos ou famílias uma ampla compreensão de todas as implicações relacionadas às doenças genéticas em discussão, as opções que a medicina atual oferece para a terapêutica ou para a diminuição dos riscos de ocorrência ou recorrência da doença genética em questão, isto é, para a sua profilaxia e eventual apoio psicoterapêutico. A prevenção secundária objetiva o estabelecimento do diagnóstico precoce, antes que a deficiência se manifeste clinicamente, que pode ser exemplificada pelo diagnóstico precoce intra-útero ou perinatal, como é o caso do exame do pezinho. Por último, a prevenção terciária, que busca limitar as conseqüências adversas da condição existente ou melhorar o nível funcional do indivíduo, representado, por exemplo, pelo trabalho de estimulação precoce desenvolvido com crianças portadoras de paralisia cerebral. Segundo autores como Frota-Pessoa (1983), Krisnky (1983) e a própria OMS (1981), a solução básica para se reduzir a incidência de portadores de DM, seria atuar nas prevenções. O grande desafio na área das deficiências mentais, hoje, são as DM congênitas de caráter herdado. Entre as causas herdadas de deficiência mental, as DM ligadas ao X(DMLX) são de grande importância, estimando-se que 20 a 25 % dos homens com DM são portadores de DMLX,
22 com alto risco de recorrência para a irmandade (Turner et al., 1996). As DMLX podem ser classificadas em dois grandes grupos (Lubs et al.,1999; Chiurazzi et al., 2004): - DMLX Sindrômica, onde a Síndrome do X frágil (FRAXA) é a mais comum, chegando a ser responsável por 2 a 6 % da DM grave e por 30 a 40% das DMLX (Gecz e Muller, 1999; Chiurazzi et al., 2004), atualmente, com a descoberta de novos genes no cromossomo X que contribuem para as DMLX, acredita-se que esta prevalência tenha diminuído para 20% (Chiurazzi et al., 2008). O gene responsável é o FMR1, localizado na região Xq27, formado por 17 éxons. A proteína codificada é a FMRP que é encontrada predominantemente em tecidos neuronais e testiculares. A principal característica dessa proteína está relacionada a sua capacidade de ligação com o RNA, participando do processo de transporte do RNAm ou com a tradução na síntese protéica (Bardoni e Mandel, 2002). É causada 99 % da vezes pela expansão do trinucleotídeo CGG localizado na região 5 UTR do gene FMR1. Na população normal a expansão de CGG varia de 5 a 50 repetições ; a pré-mutação de 50 a 200 repetições e a mutação completa com mais de 200 repetições. O fenótipo dessa Síndrome varia de acordo com o tamanho da expansão e o sexo. Os meninos com a mutação completa geralmente apresentam um DM moderado e algumas características físicas como face alongada, palato ogival e macroorquidismo pós-puberal. As meninas com mutação completa apresentam um quadro clínico mais leve. As portadoras da pré-mutação podem apresentar falência ovariana precoce e em 3% encontra-se DM. O diagnóstico laboratorial inclui a citogenética, que requer um meio de cultura deficiente em folato, para que se identifique o sítio frágil ou o exame molecular, com o estudo do gene FMR1 mediante técnicas de Southern Blot e Reação em Cadeia da Polimerase (PCR).
23 - DMLX Não Síndrômica, onde a forma mais comum é a Síndrome do FRAXE, associada a um tipo de DM leve a limítrofe e ausência de dismorfias (Gecz, 2000). O sítio frágil FRAXE é criado por mutações dinâmicas do gene FMR2, composto por 22 éxons, localizado em Xq28. A proteína codificada está relacionada a transcrição gênica (Hillman e Gecz, 2001). Suas mutações são também resultantes da expansão de uma trinca de nucleotídeos CCG, cuja expansão acima de 200 nucleotídeos leva ao silenciamento do gene FMR2. O diagnóstico laboratorial é semelhante ao FRAXA. Já foram identificados muitos outros genes envolvidos com DMLX Não Sindrômica (GDI1, OPHN1, PAK3, RSK2, IL-IRAPL1, TM4SF2, ARHGEF6, MECP2, FACL4, ATRX, etc), e a lista continua a crescer. As DM com genes localizados no cromossomo X possuem importância epidemiológica devido a sua alta freqüência relativa. Também com grande significado para o conhecimento etiológico de DM, devido o mecanismo de hemizigose no homem, que expõe mais claramente os genes no cromossomo X. Com isso, em nossos dias, existem mais genes conhecidos no cromossomo X envolvidos com a inteligência do que em todo genoma humano. Foi realizada uma avaliação genético-clínica dos deficientes matriculados na APAE de Batatais (SP) entre 2001 e 2003, incluindo posterior triagem das Síndromes do X Frágil tipo FRAXA e FRAXE por exame molecular de PCR (Giuliani,2005). Na amostra total de deficientes, foi possível estabelecer o diagnóstico etiopatogênico em 49% dos pacientes. Entre as etiologias definidas, 18,5% dos pacientes tinham doença genética; 3,2% tinham defeito isolado do SNC (Sistema Nervoso Central); 16,2% dos pacientes a causa foi ambiental; 9,5% apresentavam disfunção do SNC; 0,9% tinham DM com psicoses; entre os casos sem diagnóstico definido, 21,86% apresentavam prováveis síndromes genéticas, que permanecem em estudo. A triagem diagnóstica por PCR para as Síndromes FRAXA e FRAXE, realizada em todos os meninos sem diagnóstico, não revelou casos positivos; entretanto, em 18,75% dos meninos avaliados, o
24 heredograma mostrou padrão de herança ligado ao X, sugerindo-se que estes casos sejam submetidos ao exame de Southern Blotting para FRAXA e pesquisa de outros genes envolvidos com DMLX não sindrômica. Outro estudo com pacientes institucionalizados foi realizado na APAE de Caxias do Sul (RS) com uma amostra de 202 pacientes. O diagnóstico etiológico foi possível em 65,34% dos casos e destes, 32,15% foram de origem genética. A síndrome de Down foi detectada em 32,2% dos casos, seguida por distúrbios de herança mendeliana em 12,4%, afecções adquiridas (incluindo infecções) em 10,4% e malformações do sistema nervoso central em 4%. A alta percentagem de casos com síndrome de Down provavelmente reflete um viés de seleção (Felix et al., 1998). Atualmente é consenso que toda criança com dificuldade de aprendizado ou DM de etiologia desconhecida, com problemas de comportamento ou atraso no desenvolvimento da linguagem deva ser avaliada para a presença de mutações nos genes FMR1 e FMR2 (Oostra et al., 1993; Rosseau et al., 1991; Hecimovic et al., 2002). O método de screening indicado é o PCR com sensibilidade de 98%, uma especificidade de 98,6% e menor custo (Haddad et al., 1996; Santos et al., 2001; Tzeng et al., 2001). A genética tem-se desenvolvido muito na área médica e sua maior aplicação até o momento está no esclarecimento etiológico das deficiências congênitas. As técnicas de genética molecular trouxeram grande avanço neste sentido. Além do mapeamento de vários genes no cromossomo X envolvidos com DM, outros genes autossômicos têm sido identificados. Baseado em famílias com história de consangüinidade e DM, mapeou-se o gene da Neurotripsina PRSS12 no cromossomo 4q25 ( Molinari et al., 2002), o gene CRBN no cromossomo 3p26 (Higins et al., 2004), o gene CC21A no cromossomo 19p13 (Vanagaite et al., 2005), entre outros, caracterizando a Deficiência Mental Não Sindrômica Autossômica Recessiva. E pela verificação
25 de 2 famílias com múltiplas gerações afetadas, mapeou-se o gene MBD5 no cromossomo 2q23 (Wagenstaller et al., 2007), caracterizando a Deficiência Mental Não Sindrômica Autossômica Dominante. Permanece o desafio na área das Deficiências Mentais de mapear e clonar os milhares de genes envolvidos com o desenvolvimento do Sistema Nervoso Central (SNC). Como já estabelecido na questão das DM congênitas, a atitude mais correta seria atuar preventivamente, e para isto, é necessário que se identifique todos os genes relacionados com DM para que se desenvolvam métodos de detecção de portadores, estabelecimento correto dos riscos de recorrência, métodos de triagem populacional e diagnóstico precoce intra-útero ou in vitro.
26 2 - JUSTIFICATIVA Este trabalho é vinculado ao Projeto de avaliação clínico-genética de todos os alunos matriculados em 5 APAEs da região de Ribeirão Preto (Batatais, Altinópolis, Serrana, Cajuru, Nuporanga ), em um total de cerca de 1000 deficientes Este projeto visa estabelecer as características etiológicas da DM em nossa região. Neste trabalho específico realizamos o estudo dos indivíduos matriculados de janeiro de 2006 a dezembro de 2007 nas APAEs de Serrana e Altinópolis, em um total de 95 e 104 estudantes, respectivamente, com o intuito de definir o diagnóstico etiológico, os riscos de recorrência e as estratégias de prevenção e manejo das deficiências mentais. Ao mesmo tempo, neste trabalho, continuamos a ampliar os estudos populacionais da Síndrome do X Frágil.
27 3 OBJETIVOS 3.1 Geral: Estabelecer o diagnóstico clínico e etiológico dos alunos matriculados nas APAEs de Altinópolis e Serrana (SP). 3.2 Específico: a Verificar a freqüência relativa das etiologias ambientais e genéticas das deficiências mentais. Estabelecer os diagnósticos clínicos e etiológicos dos alunos matriculados nestas APAEs. b Realizar a triagem através das técnicas de PCR das Síndromes do FRAXA e FRAXE nestas instituições. c Levantar os casos familiais de DMLX, construindo os heredogramas, para estimarmos a prevalência de DM ligada ao X não sindrômica em nossas populações. d Realizar o Aconselhamento Genético das famílias dos alunos destas APAEs.
28 4 CASUÍSTICA E MÉTODOS Este estudo avaliou uma população de deficientes institucionalizados conjuntamente com seus familiares, nas APAEs (Associação de Pais e Amigos dos Excepcionais) de Altinópolis(SP) e Serrana(SP), onde estão matriculados 112 e 92 indivíduos, respectivamente. Esta avaliação direcionou-se para caracterizar etiologicamente as deficiências mentais nestas instituições. Para isso, foi realizado um exame genético-clínico com construção dos heredogramas e exames complementares, como o de imagem do SNC, exames metabólicos, cariótipo e análise molecular. Estes exames foram realizados na rede municipal de saúde, no Hospital das Clínicas de Ribeirão Preto (SP), no Laboratório de Citogenética Humana e Genética Molecular sob supervisão do Prof. Dr. João Monteiro de Pina-Neto (FMRP-USP). Os pacientes foram classificados quanto a seu nível de DM considerando-se o modelo atual qualitativo: - DM leve para aqueles que desenvolvem habilidades sociais, de comunicação e até profissionais, podendo ser auto-suficientes. Possuem pouco senso crítico, atenção oscilante, falhas nos processos de conceituação abstrata e atraso nos estudos, difícil ultrapassarem a 4º série. - DM moderado para aqueles com imaturidade psicomotora nos primeiros anos. São independentes nas atividades de vida diária mas necessitam de certa supervisão. Comportamento variável, muitas vezes com dificuldade de relacionamento interpessoal. Fala e linguagem comprometidas com falhas de atenção importante. Iniciam alfabetização, mas normalmente se expressam por símbolos gráficos de forma rudimentar.
29 - DM grave para aqueles com atraso no desenvolvimento psicomotor acentuado, déficit significativo na comunicação e déficit intelectual severo. Executam tarefas simples sob estrita supervisão. - DM profundo para aqueles com atraso psicomotor grave com restrição da mobilidade, comunicação eventual através de fala estereotipada e rudimentar. Incapacidade de auto-cuidados, automutilações são freqüentes e déficit intelectual profundo. Classificou-se, também, clinicamente e etiologicamente as deficiências mentais: 1) Classificação clínica - DM com anomalias múltiplas: pacientes com anomalias cromossômicas, alterações gênicas, prováveis síndromes (grupo de pacientes com características sindrômicas cujo diagnóstico genético não pode ser definido) e casos com malformações esporádicas. - DM com malformação SNC primária: pacientes com malformação SNC isolada como hidrocefalia por malformação de Chiari, defeitos de migração neuronal como a esquizencefalia, malformação de Dandy Walker, macro/microcefalia primária. - DM de causas ambientais: pacientes com diagnóstico de encefalopatia hipóxicoisquêmica (EHI), prematuridade, dano cerebral pelo uso de teratógenos na gestação, infecções congênitas, infecções pós-natais, trauma encefálico, desnutrição. - DM e disfunção do SNC: pacientes com DM e epilepsia que não possuem dismorfias e história de injúria pré ou perinatal com exame de imagem do SNC normal. - DM com psicose: pacientes com DM e distúrbio psiquiátrico.
30 -DM aparentemente puro: pacientes com DM sem malformações ou dismorfias, apresentando heredograma com padrão de herança autossômica dominante, autossômica recessiva ou ligado ao X. - DM não classificado: pacientes com DM sem causa definida, não possuem malformações ou dismorfias que justifiquem fazerem parte das prováveis síndromes. 2)Classificação etiológica - Anomalias cromossômicas: numéricas e estruturais. - Anomalias gênicas: Síndromes clássicas (autossômica dominante, autossômica recessiva, ligada os X e defeito de imprinting); DMs não-sindrômicas (provável autossômica dominante, provável autossômica recessiva e provável ligada ao X), estas baseadas em heredogramas padrões e a recentes trabalhos que identificaram vários genes no cromossomo X e em região autossômica. - Multifatorial : casos com componente genético e ambiental. - Esporádico: anomalias com risco de recorrência provavelmente desprezível. - Heterogêneo: anomalias que possuem diferentes etiologias, como a Síndrome de Moebius, que pode ser causada pelo uso de misoprostol (ambiental) ou ter padrão autossômico dominante (genético). - Ambiental: pacientes com dano cerebral confirmado por EHI, prematuridade, infecção congênita, infecção pós-natal, uso de teratógenos na gestação, trauma encefálico, desnutrição. Os exames de imagem do SNC foram realizados em pacientes com história clínica de prematuridade, EHI ou infecção congênita; presença de microcefalia ou macrocefalia no exame físico; suspeita de craniosinostose; alterações no exame neurológico; DM ou ADNPM sem causa definida após outras investigações.
31 O exame de cariótipo foi realizado primeiramente nos casos com anomalias múltiplas e em seguida nos casos sem diagnóstico etiológico definido. A análise molecular foi realizada por PCR para FRAXA, FRAXE. Os pacientes foram previamente selecionados de acordo com o seguinte critério: FRAXA e FRAXE pacientes do sexo masculino com deficiência mental de etiologia desconhecida. O Screening do FRAXA foi feito conforme o protocolo de Haddad et al. (1996), em que utilizou-se três primers F, Eag-U, Eag-L (tabela 1), 100 ng de DNA do paciente que foi amplificados num volume total de 25μl (50 mm de KCl, 10mM de TRIS-HCl, 0,1% de Triton X- 100, 2 mm de MgCl 2, 10% de dimetilsulfoxitide, 200 μm de cada datp, dctp e TTP, 50 mm de dgtp, 150 mm de 7-deaza-dGTP, 0,8 μm do primer Eag-L, 0,3 μm do primer Eag-U, 0,2 Μm do primer Eag-F). A técnica de PCR foi realizada com 35 ciclos com 1minuto e 30 segundos para a desnaturação do DNA a 94ºC, 1 minuto para o anelamento a 67ºC e 2 minutos para a extensão a 72ºC, com uma extensão final de 10 minutos. O DNA dos pacientes e o marcador de peso molecular foram separados em um gel de poliacrilamida a 5%, numa corrida de 100W-117W por 1 hora e 40 minutos.
32 Tabela 1 - Primers utilizados para o método de PCR para detecção de FRAXA FRAXA (protocolo de Haddad et al, Hum Genet)* f 5 agc ccc gca ctt cca cca cca gct cct cca 3 Eag-U 5 cga cct gtc acc gcc ctt cag cct tcc 3 Eag-L 5 cgc tgc ggg tgt aaa cac tga aac cac gtc 3 O Screening do FRAXE foi feito conforme o protocolo de Santos et al. (2001), em que utilizou-se três primers 603, XE1, XE2 (tabela 2), 150 ng de DNA do paciente que foram amplificados num volume total de 25μl (50 mm de KCl, 10mM de TRIS-HCl,, 1,6 mm de MgCl 2, 10% de dimetilsulfoxitide (DMSO), 200 μm de cada datp, dctp e TTP, 50 mm de dgtp, 150 mm de 7-deaza-dGTP, 0,2 μm do primer XE1, 0,6 μm do primer XE2, 0,4 μm do primer 603). A técnica de PCR foi realizada com 30 ciclos com 1minuto e 30 segundos para a desnaturação do DNA a 95ºC, 1 minuto para o anelamento a 65ºC e 2 minutos para a extensão a 72ºC, com uma extensão final de 10 minutos. O DNA dos pacientes e o marcador de peso molecular foram separados em um gel de policrinamina a 5%, numa corrida de 100W-117W por 1 hora e 40 minutos.
33 Tabela 2 - Primers utilizados para o método de PCR para detecção de FRAXE FRAXE (protocolo de Santos et al, Hum Mutation) XE1 _ 5 ggc ggg cgg ccc agc ccg cct gag cc 3 XE2 _ 5 gcc ctc ccg ccc agc taa aag tgt ccg gg _ 5 cct gtg agt gtg taa gtg tgt gat gct gcc g 3 Os pacientes selecionados para cariótipo e exame molecular assinaram o Termo de Consentimento Livre Esclarecido aprovado pelo Comitê de Ética em Pesquisa do HCFMRP- USP e pelo CONEP (n 12715/2006).
34 5 RESULTADOS 5.1 GERAL A avaliação foi realizada nos 205 pacientes com exclusão de 5 pacientes (total de 200 pacientes) devido apresentarem somente deficiência física ou deixarem a instituição antes do término das investigações. A idade variou de 0 a 69 anos com 114 (57%) pacientes do sexo masculino e 86 (43%) do sexo feminino (tabela 3). A consangüinidade foi encontrada em 3 famílias (1,5%), sendo 2 delas pertencentes ao grupo das doenças autossômicas recessivas. Tabela 3: Distribuição da amostra segundo o gênero. M F TOTAL APAE ALTINÓPOLIS n % n % 57 53, ,8 107 APAE SERRANA TOTAL CLASSIFICAÇÃO QUANTO AO NÍVEL DE DM Os pacientes foram classificados quanto a seu nível de DM conforme a descrição do DMS IV realizada por equipe multidisciplinar. Observou-se que 6% apresentaram DM leve, 48,5% DM moderada, 22,5% DM grave e 15% DM profunda. Em 8% dos pacientes não foi possível estimar o grau de DM devido a faixa etária, classificando-se, assim, como ADNPM (tabela 4).
35 Tabela 4: Classificação quanto ao nível de DM. LEVE MODERADA GRAVE PROFUNDA ADNPM TOTAL n % n % n % n % n % APAE 8 7, ,5 107 ALTINÓPOLIS APAE SERRANA 93 TOTAL , , ADNPM = atraso do desenvolvimento neuropsicomotor. 5.3 CLASSIFICAÇÃO CLÍNICA E ETIOLÓGICA DAS DMs De acordo com a avaliação genético-clínica dos 200 pacientes das APAEs de Serrana e Altinópolis, classificou-se as deficiências mentais clinicamente e etiologicamente. As características dos pacientes quanto a classificação clínica e etiológica encontram-se na Tabela 5 e 6, respectivamente. Classificou-se, clinicamente, 23% dos pacientes com DM com anomalias múltiplas (37 casos de Síndromes Genéticas, 7 casos de Prováveis Síndromes, 1caso de Lipomatose Encefalocutânea e 1 caso de Seqüência de Moebius), 4% DM com malformação do SNC isolada (1 caso de Displasia Septo-Óptica, 1 casi de Esquizencefalia, 4 casos de Malformações de Dandy Walker e 1caso de Microcefalia), 3% DM e erros inatos do metabolismo (4 casos de fenilcetonúrias e 2 casos de Síndrome de Smith-Lemli-Opitz), 42% com DM de causa ambiental ( Figura 4 ), 1% DM e disfunção do SNC ( 2 casos de DM e epilepsia), 7% DM aparentemente puro [8 casos de DM não sindrômico com provável herança autossômico
36 dominante, 1 caso com padrão autossômico recessivo e 4 casos com padrão ligado ao X recessivo (ANEXO)] e 20% DM não classificado (Figura 1). As características dos pacientes classificados como prováveis síndromes encontram-se na Tabela 7. Tabela 5: Classificação Clínica das Deficiências Mentais por APAE. GRUPOS CLÍNICOS SERRANA ALTINÓPOLIS TOTAL n % n % DM com anomalias múltiplas 18 19, DM com malformação SNC isolada 6 6, DM e erros inatos do metabolismo 4 4, DM e disfunções do SNC DM e psicoses DM de causa ambiental DM aparentemente puro padrão autossômico dominante ,5 8 padrão autossômico recessivo padrão ligado ao X recessivo DM não classificado 19 20, ,5 40 TOTAL
37 20% CLASSIFICAÇÃO CLÍNICA 23% DM com malformações congênitas múltiplas. DM com malformação isolada do SNC. DM de causa ambiental. 7% DM e erros inatos do metabolismo. 1% 3% 42% 4% DM e disfunção SNC. DM aparentemente puro. DM não classificado. Figura 1: Classificação Clínica Geral das Deficiências Mentais em porcentagem.
38 Tabela 6: Classificação Etiopatogênica das Deficiências Mentais. SERRANA ALTINÓPOLIS TOTAL Anomalias cromossômicas n % numéricas estruturais Anomalias gênicas Síndromes clássicas autossômica recessiva autossômica dominante ,5 ligada ao X defeito de imprinting ,5 DM não-sindrômica provável autossômica recessiva ,5 provável autossômica dominante provável ligada ao X Multifatorial ,5 Esporádico / desconhecido ,5 Heterogêneo ,5 Causas Ambientais prematuridade EHI ,5 infecção congênita ,5 infecção pós-natal ,5 teratógeno ,5 seqüela de TCE ,5 85 DM indefinido provável síndrome ,5 não identificado TOTAL
39 Quanto a etiologia, 28% dos pacientes apresentaram DM de causa genética (14% anomalias cromossômicas e 14% anomalias gênicas), 42% de causa ambiental, 4% heterogêneo, 2% esporádico, 1% multifatorial e 23% indefinido (Figura 2). A descrição dessas etiologias encontra-se na Tabela 8 observando-se entre as anomalias cromossômicas, 24 casos de Síndrome de Down e 4 casos de anomalias estruturais [46,XY, add (11) (p15); 46,XX, add (3) (q29); 2 casos 46,XX, add (22) (q13)], e entre as anomalias gênicas, 12 casos de síndrome clássica autossômica recessiva [4 casos de fenilcetonúrias, 2 casos de Síndromes de Smith- Lemli-Opitz, 1 caso de Síndrome de Seckel (FOTO A), 1 caso de Síndrome de Carpenter (FOTO B), 1 caso de Síndrome de Cohen (FOTO C), 2 casos de Síndromes de Usher, 1 caso de Síndrome de Pitt-Rogers (FOTO D)], 1 caso de síndrome clássica autossômica dominante (microcefalia autossômica dominante), 2 casos de síndrome clássica ligada ao X [Síndrome do X-frágil (FOTO G) e Síndrome de DM ligado ao X, macroorquidismo, face acromegálica (FOTO E)] e 1 caso de defeito de imprinting [Síndrome de Beckwith Wiedemann (FOTO F)]. Os casos de síndromes clássicas sem exame especial confirmatório foi discriminado na Tabela 9. Cada APAE foi classificada separadamente para análise das diferentes etiologias em cada uma delas. Na APAE de Serrana encontrou-se 24% de origem genética, 45% ambiental e 22,5% ficaram sem diagnóstico. Na APAE de Altinópolis observou-se 32,8% de etiologia genética, 40% ambiental e 24,5% indefinidos (Figura 3).
40 CLASSIFICAÇÃO ETIOLÓGICA 1% 4% 2% 23% 28% genético. ambiental. esporádico. heterogêneo. multifatorial. indefinido. 42% Figura 2: Classificação etiológica das Deficiências Mentais.
41 Tabela 7: Sete casos classificados como prováveis síndromes genéticas. N NOME CARACTERÍSTICAS QI CARIOTIPO IMAGEM SNC 1 A.S.S. face com dismorfias, clinodactilia 5º dedos, pseudocampto de 4 dedo D, frouxidão ligamentar, hirsutismo, halux valgo M normal CT e RM normais OUTROS HDx Sd. de Microdeleção? 2 D.A.M face hipotônica, frontal amplo, filtro naso labial longo,pectus escavatum frouxidão ligamentar, voz anasalada 3 E.A.S. face simiesca, microcefalia,hipotelorismo, lábios finos,hipertricose, articulações proeminentes M normal RM normal ECO, ENMG, EIM, FRAXA normais M normal CT normal ECO, RX mãos, pés, ossos longos normais Sd. Aaskorg? Sd. Trico- Rino- Falangeana tipo II? 4 H.D.M prognatismo, hipertelorismo, frouxidão ligamentar, dedos dos pés e mãos afilados, hipertricose,estereotipias, voz fina infantilizada G normal CT cisto de Dandy Walker FRAXA e FRAXE normais Sd. Coffin- Lowry? 5 J.L.F. microcefalia, retrognatia, estrabismo, nariz longo, discreto algargamento de polegares G normal CT normal e RM com subst. branca EIM normal Sd. Rubinstein Taybi? 6 M.A.S. microbraquicefalia, hipertelorismo, hipoplasia malar, filtro naso-labial curto, autoagressividade, marcha com base alargada G normal CT desproporção crânio-face Sd. Angelman? 7 R.F.L. comportamento autista, estereotipias com as mãos, autoagressividade, telecanto P normal RM discreta atrofia subcortical bifrontal BERA e EIM normais Sd. Rett?
42 Tabela 8: Descrição de cada etiologia. Anomalias cromossômicas n=28 foto Síndrome de Down 24 46,XY,add(11)(p15) 1 G 46,XX,add(3) (q29) 1 H 46XX,add(22)(q13) 2 I/J Anomalias gênicas n=29 Síndromes Clássicas Autossômica Recessiva n=12 Fenilcetonúria 4 Smith-Lemli-Optiz tipo 1 2 Síndrome de Seckel 1 A Síndrome de Carpenter 1 B Síndrome de Cohen 1 C Síndrome de Usher 2 Síndrome de Pitt-Rogers 1 D Autossômica Dominante n=1 Microcefalia autossômica dominante 1 Ligada ao X n=2 Síndrome do X-frágil 1 N DM ligado ao X, macroorquidismo, face acromegálica 1 E Defeito de imprinting n=1 Síndrome de Beckwith-Wiedemann 1 F Multifatorial n=1 Malformação de Chiari tipoii 1 Esporádico n=3 Lipomatose encéfalo-cutânea 1 M DM e epilepsia 2 Heterogêneo n=7 Displasia septo-óptica 1 Esquizencefalia 1 Malformação de Dandy Walker 4 L Seqüência de Moebius 1
43 Tabela 9: Descrição das síndromes clássicas com diagnóstico clínico. NOME DIAGNÓSTICO FENÓTIPO + EXAMES R.B.S. Síndrome de Seckel L.M.S. T.L.G. Síndrome de Carpenter Síndrome de Cohen microcefalia (PC=45 cm) baixa estatura proporcionada ( Est= 1,35) nariz grande com raiz nasal alta região maxilar proeminente má oclusão dentária orelhas proeminetes aumento espaço entre 1º e 2 artelhos cranioestenose braquicefalia frontal amplo olhos proeminentes obesidade de tronco clinodactilia 5º quirodáctilo braquidactilia criptorquidia e hipospadia distal hipotonia obesidade estreitamento bitemporal incisivos superiores proeminentes filtro naso labial curto palato alto e estreito quirodáctilos longos. afilamento falanges distais Cariótipo nl ECO nl CT crânio com megacisterna magna Cariótipo nl Audiometria nl Exame oftalmológico nl RX crânio com fechamento de suturas coronais CT crânio com dilatação ventricular CT nl Exame oftalmológico com hipemetropia T.M.S. Síndrome de Pitt- Rogers microcefalia baixa estatura face característica com olhos proeminentes hipoplasia de face média telecanto filtro naso labial curto lábios finos helix e anti-helix hipoplásicas pit pré-auricular E mãos pequenas Cariótipo nl CT crânio com atrofia cortical




44 Tabela 9: Continuação das síndromes clássicas com diagnóstico clínico. NOME DIAGNÓSTICO FENÓTIPO + EXAMES A.O.P. Síndrome de DM ligado ao X, macroorquisdismo, face acromegálica prognatismo região supra-orbital proeminente sombrancelhas grossas nariz longo orelhas proeminentes macroorquidismo Cariótipo nl RM encéfalo nl FRAXA e FRAXE (-) V.M.S. Síndrome de Beckwith Wiedemann hipoglicemia e hipocalcemia ao nascimento macroglossia hemihipertrofia hérnia umbilical Ultra-som abdominal nl ECO nl FOTO A: R.B.S. SÍNDROME DE SECKEL.




45 FOTO B: L.M.S. SÍNDROME DE CARPENTER RX de crânio perfil evidenciando fechamento da sutura coronal e impressões digitiformes RX de mão com deformidade no 5 quirodáctilo






46 FOTO C: T.L.G. SÍNDROME DE COHEN FOTO D: T.M.M.S. SÍNDROME DE PITT-ROGERS


47 FOTO E: A.O.P SÍNDROME DM LIGADO AO X, MACROORQUIDISMO, FACE ACROMEGÁLICA FOTO F: V.M.S. SÍNDROME DE BECKWITH WIEDEMANN
48 % ,8 COMPARAÇÃO ETIOLÓGICA ENTRE APAEs ,5 1,8 2 0, ,5 24,5 genético ambiental heterogêneo esporádico multifatorial não classificado APAE SERRANA APAE ALTINÓPOLIS Figura 3: Classificação etiológica da APAE de Serrana e Altinópolis separadamente. Dentro das causas ambientais, as etiologias foram diversas. A encefalopatia hipóxicoisquêmica (EHI) compreendeu 39% dos casos (Serrana 31% e Altinópolis 46,5%), seguida da prematuridade com 33% (Serrana 36% e Altinópolis 30%), infecção congênita 13% (Serrana 14% e Altinópolis 12%), infecção pós-natal 8% (Serrana 9,5% e Altinópolis 7%), uso de teratógenos 6% (Serrana 7% e Altinópolis 4,5%) e seqüela traumatismo crânio-encefálico (TCE) 1% (Serrana 2,5% e Altinópolis 0%) (Figura 4 e 5).
49 % 33 CAUSAS AMBIENTAIS TOTAL prematuridade EHI infecção congênita infecção pós-natal teratógeno seqüela TCE Figura 4: Distribuição das freqüências das etiologias no grupo das causas ambientais. CAUSAS AMBIENTAIS % APAE SERRANA APAE ALTINÓPOLIS prematuridade EHI infecção congênita infecção pós-natal teratógeno seqüela TCE Figura 5: Distribuição das freqüências das causas ambientais por APAE.
50 Entre as causas ambientais, a EHI e a prematuridade por sua relevância nesta amostra, foram distribuídas pela década em que foram geradas com o objetivo de se identificar a evolução temporal desses problemas segundo a freqüência de cada causa (Figura 6). Observou-se apenas casos de EHI (6%) na década de 60; na década de 70 encontrou-se 12% de EHI e 3,5% de casos de prematuridade; 27% e 21,5% na década de 80, respectivamente; 30% e 46,5% na década de 90; 25% e 28,5% a partir do ano Considerou-se, entre os 33 casos de EHI, que a hipóxia prénatal foi responsável por 12% dos casos, que a perinatal correspondeu a 79% e 3% não foi possível identificar. Quanto ao local de nascimento, obteve-se que 21% dos casos nasceram em hospitais de alta complexidade, 66% em hospitais de baixa complexidade e 9% o parto foi domiciliar. Verificou-se que 40% destes casos permaneceram internados até 1 mês de vida, 12% de 1 a 2 meses, 9% superior a 2 meses, 12% não foi possível identificar e 27% não necessitaram de internação (Tabela 10). Entre os 28 casos de prematuridade, 53% dos casos tiveram peso ao nascer menor ou igual a 1500g, 40% apresentaram peso maior que 1500g e 7% não foram identificados; 64% nasceram em hospitais de alta complexidade, 32% em hospitais de baixa complexidade e 4% nasceram em domicílio; 14% ficaram internados até 1 mês de vida, 43% de 1 a 2 meses, 29% superior a 2 meses e 14% indefinido (Tabela 11).
51 % dec.60 dec.70 dec.80 dec.90 >2000 EHI prematuridade total por faixa etária Figura 6: Distribuição por faixa etária das duas causas ambientais principais. Tabela 10: Características dos casos de encefalopatia hipóxico-isquêmica HIPÓXIA n % LOCAL DE n % INTERNAÇÃO n % NASCIMENTO perinatal Hospital de alta 7 21 até 1 mês complexidade pré-natal 4 12 Hospital de baixa a 2 meses 4 12 complexidade indefinido 3 9 Domiciliar 3 9 > 2meses 3 9 indefinido 4 12 não internaram 9 27 TOTAL Tabela 11: Características dos casos de prematuridade PESO n % LOCAL DE n % INTERNAÇÃO n % NASCIMENTO 1500g Hospital de alta até 1 mês 4 14 complexidade >1500g Hospital de baixa a 2 meses complexidade indefinido 2 7 Domiciliar 1 4 > 2 meses 8 29 indefinido 4 14 TOTAL
52 As outras causas ambientais como a infecção congênita, observou-se um predomínio de toxoplasmose (64%), seguida de rubéola (9%) e citomegalovirose (9%). Houveram 2 casos (18%) de infecção congênita em que não foi possível identificar o agente, sendo o diagnóstico baseado na história clínica e exame de imagem com calcificações patológicas (Tabela 12), a descrição de cada paciente encontra-se na tabela 13. Os vários tipos de meningite foram as causas de todas infecções pós-natais: meningite por Haemophilus B (14%), meningite pneumocócica (14%), meningite meningocócica (29%), meningite viral (14%), meningite não identificada (29%) [Tabela 12]. A descrição desses pacientes encontra-se na tabela 14. Nesta amostra o uso de álcool na gestação foi o único teratógeno encontrado (Tabela 12). Foram identificados 5 casos baseados em protocolo específico (Tabela 15). Tabela 12: Outras etiologias ambientais observadas nesta amostra. Infecção congênita n % toxoplasmose 7 64 rubéola 1 9 citomegalovírus 1 9 outros Infecção pós-natal meningite Haemophilus B 1 14 meningite pneumocócica 1 14 meningite meningocócica 2 29 meningite viral 1 14 meningite não identificada Teratógeno efeitos fetais do álcool 5 100
53 Tabela 13: Descrição dos pacientes com diagnóstico de infecção congênita. NOME QI DX SOROLOGIA PACIENTE T.M.A. G Toxoplasmose IgG = 1/4000 IgM - I.M.S. P Toxoplasmose IgG = 1/4000 IgM - EXAMES CLÍNICA OBSERVAÇÕES CT crânio com calcificações patológicas F.O. nl EEG com epilepsia focal sintomática CT crânio com calcificações patológicas F.O. coriorretinite bilateral G.M.R. G Toxoplasmose - CT crânio com calcificações patológicas F.O. lesão em fenda bilateral G.V.S. M Toxoplasmose IgG / IgM + CT crânio com calcificações patológicas F.O. coriorretinite R.N. P Toxoplasmose IgG / IgM + CT crânio com calcificações cerebrais CT coluna com calcificações no interior do corpo medular (T12) F.O. sinéquias posteriores L.D. P Toxoplasmose IgG / IgM + RX crânio com microcefalia F.O. coriorretinite Microcefalia Espasticidade > E Crises convulsivas desde 4 anos de idade Microcefalia Tetraespasticidade Crises convulsivas desde o 1º ano de vida Microcefalia Espasticidade > membros inferiores Surdez bilateral Microbraquicefalia Estrabismo Nistagmo horizontal Microcefalia Enoftalmia Hipotonia global Microcefalia Espasticidade > membros inferiores Amaurose bilateral História ADNPM História ADNPM Ultra-som gestacional com hepatomegalia e alteração SNC História de parto prematuro e ADNPM História ADNPM História ADNPM História ADNPM e hepatomegalia ao nascimento D.G.L. M Toxoplasmose IgG = 1/4000 IgM - CT crânio com calcificações patológicas F.O. coriorretinite Microcefalia Nistagmo horizontal e estrabismo convergente História ADNPM D.S.M. M Rubéola - CT crânio nl Exame oftalmológico com catarata congênita B.M.S. G CMV PCR + (2 anos) RM com dilatação ventricular e atrofia cerebral BERA hipoacusia M.C.S. G?? CT com calcificações patológicas F.O. coriorretinite L.R.S. M?? CT crânio com calcificações patológicas Microcefalia Alteração visão Nistagmo Crises convulsivas desde 2 anos de idade Microcefalia Hipoacusia Microcefalia Nistagmo horizoantal Estrabismo convergente Microcefalia Hipotonia Rubéola detectado no 6 mês de gestação História de parto prematuro e ADNPM História ADNPM História de parto domiciliar e ADNPM História de parto prematuro e ADNPM
54 Tabela 14: Descrição dos pacientes com diagnóstico de infecção pós-natal. NOME QI DX EXAME IMAGEM SNC CLÍNICA L.A.B. L Meningite meningocócica com 19 dias CT com coleção subdural bilateral (empiema) e atrofia cortical difusa Microcefalia M.S.S. A D N P M Meningite meningocócica com 3 dias J.G.O. G Meningite pneumocócica com 1 mês J.P.S. M Meningite Haemophilus B com 1 ano A.J.S. P Meningite viral com 20 dias A.J.F. G Meningite não identificada com 5 meses D.S.S. P Meningite não identificada com 1 ano RM com acentuada redução do parênquima cerebral com encefalomalácea multicística RM com hemiatrofia cerebral D com sinais de redução volumétrica da região frontal E CT nl CT com leucomalácea periventricular multicística RM com sinais de perda volumétrica cerebral RM com áreas multifocais de atrofia com gliose adjacente Microcefalia Hipotonia global Crises convulsivas Microcefalia Hipotonia com hemiparesia à E Normocefálico Reflexos tendíneos e tônus sem alterações Microcefalia Tetraparesia espástica Crises convulsivas Microcefalia Tetraparesia espástica Microcefalia Crises convulsivas
55 Tabela 15: Descrição dos pacientes com diagnóstico de efeitos fetais do álcool. Nome Exposição materna confirmada ao álcool Retardo de crescimento pré ou pós natal Anomalias menores Anomalias cerebrais estruturais Anomalias cognitivas ou comportamentais W.A.E.C. * Clinodactilia 5 QD D.C. * * Microcefalia, raiz nasal baixa, lábio superior fino L.C.S.G. * Microcefalia, filtro nasolabial apagado, lábio superior fino, prognatismo L.G.P. * * Microcefalia, lábio superior fino, filtro naso-labial apagado D.C.C. * * Microcefalia, lábio superior fino, filtro naso-labial apagado, epicanto Agenesia de septo pelúcido e alteração de corpo caloso Agenesia de corpo caloso * * * * * Para cada grupo etiológico foi estabelecido o risco de recorrência (risco baixo < 5%, risco médio 5 a 10% e risco alto > 10%) com a finalidade de se estabelecer o melhor manejo desses pacientes e realizar o aconselhamento genético para as famílias. - Grupo das anormalidades cromossômicas numéricas: Síndrome de Down trissomia livre com mãe jovem (n = 2) risco baixo
56 Síndrome de Down trissomia livre com mãe > 30 anos (n = 22) risco é dependente da idade materna (idade acima 30 anos risco de 1% e acima de 40 anos risco de 2%) - Grupo das anormalidades cromossômicas estruturais 46,XY,add(11)(p15) = não identificado (não foi possível realizar o cariótipo do pai) 46,XX,add(3) (q29) = desprezível (cariótipo dos pais normais) 46XX,add(22)(q13) = risco alto [pai com cariótipo 46,XY;t(18;22) (p11.3;q13)] - Grupo das anomalias gênicas / síndromes clássicas Autossômicas recessivas = risco alto Autossômicas dominantes = risco alto Ligadas ao X = risco alto Defeito de Imprinting = não identificado (não foi possível realizar exame molecular para Síndrome de Beckwith Wiedemann) - Grupo das anomalias gênicas / DM não sindrômica Prováveis autossômicas recessivas = risco alto Prováveis autossômicas dominantes = risco alto Prováveis ligadas ao X = risco alto - Grupo de causa multifatorial Malformação de Chiari tipo II = risco médio
57 - Grupo de causa esporádica Lipomatose encéfalo-cutânea = risco baixo DM e epilepsia = não identificado - Grupo de causa heterogênea Displasia septo-óptica = baixo Esquizencefalia = baixo Malformação de Dandy Walker = baixo Seqüência de Moebius = não identificado - Grupo de causa ambiental Dependente de se afastar o fator causal 5.4 CLASSIFICAÇÃO ETIOLÓGICA E NÍVEL DM Na Figura 6 o nível de inteligência foi distribuído por grupo etiopatogênico. Os pacientes com anomalias cromossômicas, 93% foram classificados como DM de leve a moderado e apenas 7% com DM de grave a profundo. As anomalias gênicas, 69% foram DM de leve a moderado, 27,5% DM grave a profundo e 3,5% foram classificados como ADNPM. Aqueles com prováveis síndromes, 43% tinham DM de leve a moderado e 57% dm grave a profundo. O grupo de causa ambiental apresentou 36,5% de DM leve a moderado, 54% de DM grave a profundo e 9,5% com ADNPM.
58 Classificação etiológica e nível de DM % DM leve-moderado DM grave-profundo ADNPM anomalias cromossômicas anomalias gênicas provável síndrome ambiental Figura 6: Demonstração do nível de inteligência divididos por grupo etiopatogênico. 5.5 EXAMES COMPLEMENTARES Nesta amostra de 200 pacientes foram realizados 145 exames de imagem do SNC e foi observado 100 (69%) exames alterados. O cariótipo foi realizado em 91 pacientes, sendo 28 (30%) alterados. O exame molecular para FRAXA e FRAXE foi realizado em 35 pacientes do sexo masculino e 1 paciente (2,8%) foi diagnosticado como Síndrome do X - frágil. Os 17 exames para erros inatos do metabolismo realizados, 6 (35%) apresentaram alterações (Tabela 16).
59 NORMAL (%) ALTERADO (%) TOTAL EXAME DE IMAGEM 45 31% % 145 CARIÓTIPO 63 70% 28 30% 91 FRAXA/FRAXE 34 97,2% 1 2,8% 35 EIM 11 65% 6 35% 17 Tabela 16: Classificação dos exames complementares CITOGENÉTICA Foram realizados 63 cariótipos nesta amostra sendo detectados 28 (30%) com anormalidades cromossômicas (85,7% de anormalidades numéricas e 14,2% de anormalidades estruturais), mostrados nas Tabelas 16 e 17. Para as anormalidades numéricas, o exame citogenético possibilitou confirmar a suspeita clínica de 24 casos de Síndrome de Down e investigar o risco de recorrência, que foi baixo, pois todos apresentaram trissomia livre. Entre as 4 anormalidades estruturais, a citogenética foi decisiva para os diagnósticos. O caso correspondente ao cariótipo 46,XY,add(11)(p15) é filho único de um casal não consangüíneo, possui história de ADNPM que evoluiu para uma deficiência mental grave. Suas manifestações clínicas compreenderam: dolicoturricefalia, fendas palpebrais para baixo, sinofre, telecanto, raiz nasal alargada, orelhas proeminentes, sulco naso-labial pouco marcado, clinodactilia de 5 quirodáctilos, sulco sacral, criptorquidia e hérnia inguinal bilateral (FOTO G). O exame citogenético da mãe deste caso não apresentou anormalidades, mas não foi possível estimar o risco de recorrência devido o pai não estar disponível para o exame. O caso cujo exame de cariótipo evidenciou alterações estruturais no cromossomo 3 [46,XX,add(3) (q29)] é a terceira
60 filha de um casal não consangüíneo, apresentando história de hipotonia ao nascimento, malformação cardíaca (comunicação interatrial e comunicação interventricular) que necessitou de correção cirúrgica no primeiro ano de vida, ADNPM que evoluiu para uma DM profunda. Seu exame dismorfológico mostrou estigmas da Síndrome de Cornélia de Lange como microcefalia, hipertricose, sinofre, baixa implantação de cabelos, cílios longos, narinas antevertidas, sindactilia de 4 e 5 pododáctilos e sindactilia parcial de 2 e 3 pododáctilos (FOTO H). O risco de recorrência foi desprezível devido constatar ser um evento novo, os pais apresentaram cariótipos sem alterações. Os outros dois casos de anomalias estruturais apresentaram a mesma alteração no exame citogenético [46,XX,add(22)(q13)], são duas irmãs, filhas de um casal não consangüíneo. A primeira filha apresentou ADNPM discreto e evoluiu para uma DM moderada. Não foram detectadas anomalias maiores nesta paciente que possui fácies muito semelhante a mãe (frontal amplo, epicanto, filtro naso-labial longo e marcado, orelhas rodadas posteriormente). A segunda filha possui ADNPM e apenas anomalias menores como estrabismo convergente bilateral, epicanto, filtro naso-labial longo e marcado com narinas antevertidas (FOTO I/J). O pai destas pacientes apresentou cariótipo 46,XY,t(18;22) (p11.3;q13), mostrando que há risco de recorrência. Tabela 17: Descrição das anomalias cromossômicas detectadas pela citogenética. ANOMALIAS CROMOSSÔMICAS N=28 % NUMÉRICAS 47,XX + 21 / 47,XY ,7 ESTRUTURAIS 46,XY, add(11) (p15) 1 46,XX, add(3) (q29) 1 46,XX, add(22) (q13) ,2

")


61 FOTO G: CARIÓTIPO 46,XY, add(11) (p15) FOTO G: J.V.S.

")

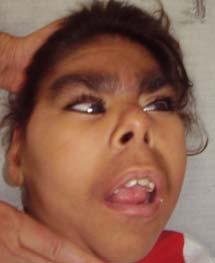

62 FOTO H: CARIÓTIPO 46,XX add(3) (q29) FOTO H: D.F.S. FOTO H: PÉ DE D.F.S

")



63 FOTO I: CARIÓTIPO 46,XX add (22) (q13) FOTO I M.F.R. FOTO I J.B.R.

")
64 FOTO J: CARIÓTIPO DO PAI DE M.F.R. e J.B.R. 46,XY;t(18;22) (p11.3;q13)
65 5.5.2 EXAME DE IMAGEM DO SISTEMA NERVOSO CENTRAL O exame de imagem do SNC (ultra-som de crânio, tomografia computadorizada de crânio e ressonância magnética de encéfalo) foi realizado em 145/200 pacientes e 69% dos exames estavam alterados (Tabela 17). Destes, a maioria (75%) das alterações foram para confirmar o diagnóstico clínico, por exemplo, nos casos de história de injúria neonatal, infecção congênita, infecção pós-natal, uso de teratógeno; 8% definiram o diagnóstico (Tabela 18), como nos casos de malformação do SNC isolada; 17% dos exames alterados foram inconclusivos (Figura 8). Tabela 18: Exame de imagem decisivo para o diagnóstico de DM NOME QI EXAME DE IMAGEM FOTO K.B.S. P Esquizencefalia S.R.Z. ADNPM Displasia septo-óptica P.F.B.E. M Malformação de Dandy Walker B.H.P.S. M Malformação de Dandy Walker A.M.S. M Malformação de Dandy Walker V.R.S.A. ADNPM Malformação de Dandy Walker L K.C.M. G Malformação de Chiari II L.A.F. G Lipomatose encéfalo-cutânea M
66 CARACTERÍSTICAS DO EXAME DE IMAGEM SNC não definiram 17 confirmaram diagnóstico definiram diagnóstico 8 75 TOTAL DE EXAME ALTERADO (N=100) Figura 8: Características dos exames de imagem quanto a definição diagnóstica de imagem.
 e o cisto de fossa posterior (asteristico), vistos em A, E e F.")
67 Ressonância Magnética de encéfalo mostrando em A uma imagem axial T2, em B uma imagem axial T2 com atenuação de liquido livre (FLAIR), em C e D imagens T1 axial, uma imagem T1 coronal em E e uma sagital em F. Note a fenda no cerebelo (seta preta) e o cisto de fossa posterior (asteristico), vistos em A, E e F. Em B e D nota-se perda da substância branca periventricular e aproximação da substância cinzenta cortical dos ventrículos (seta branca), sugerindo leucomalácia periventricular. Em B, D, E e F observase ausência do corpo caloso com formação de banda de Probst (cabeça de seta). FOTO L


68 Imagem de Ressonância magnética com imagem ponderada em T2, axial, em A, T1 axial em B e T1 axial, com subtração de gordura em C. Em D, E e F as imagens são ponderadas em T1, após injeção de contraste endovenoso, nos planos sagital, axial e coronal, respectivamente. A seta branca aponta um lipoma na porção superior da cisterna do ângulo ponto cerebelar, vista em A, B, C, D e F. Note que em C, com a subração da gordura a imagem perde o brilho, da mesma maneira que o tecido gorduroso subcutâneo. A criança apresenta também um cisto aracnóide temporal direito (asterisco), associado com atrofia do lobo temporal. Todo o restante do hemisfério direito é atrófico, com redução de espessura cortical, notando-se reforço intenso pós-contraste, de um hemangioma veno-capilar (pequenas setas brancas em E e F). FOTO M
69 5.5.3 EXAME PARA ERROS INATOS DO METABOLISMO Os exames para erros inatos do metabolismo (EIM) não foram realizados de rotina neste estudo. Os casos foram selecionados de acordo com a presença de características sugestivas para EIM como: história de regressão no DNPM, crises convulsivas de difícil controle, exame dismorfológico compatível, etc. Obteve-se uma amostra de 17 pacientes, e 6 (35%) apresentaram alterações (Tabela 17). O diagnóstico de fenilcetonúria foi realizado em 4 pacientes, todos não possuiam a triagem neonatal (exame do pezinho) e tiveram história semelhante de regressão no DNPM a partir dos 2 anos de vida. Dois deles ficaram sem tratamento até mais de 20 anos, desenvolvendo uma DM mais severa. Os outros casos com EIM foram de 2 irmãos, filhos de casal consangüíneo com fenótipo característico (microcefalia, ptose palpebral, narinas antevertidas, sindactilia cutânea de 2 e 3 pododáctilos, membros hipotróficos) de Síndrome de Smith-Lemli- Opitz, confirmado por exame específico de dosagem de 7-dehidrocoleterol, um substrato da via de síntese do colesterol que encontra-se com valor aumentado devido a deficiência da enzima 7- dehidrocolesterol redutase (Tabela 19).
70 Tabela 19: Pacientes com exame EIM positivo. NOME QI EIM OBSERVAÇÕES J.L.N.F. G fenilcetonúria 4 filho de casal não consangüíneo com história de regressão DNPM a partir dos 2 anos. Diagnóstico com 33 anos, apresentando DM sem dismorfias e pele clara. Possui irmão afetado que faz parte desta amostra. L.N.F. G fenilcetonúria 3 filho de casal não consangüíneo com história de regressão DNPM a partir dos 2 anos. Diagnóstico com 39 anos, apresentando DM sem dismorfias e pele clara. Possui irmão afetado que faz parte desta amostra. G.A.A.V. M fenilcetonúria 2 filho de casal não consangüíneo com história de regressão DNPM sem idade precisa. Diagnóstico com 3 anos com exame físico sem dismorfias. V.D.A. M fenilcetonúria 2 filho de casal não consangüíneo com história de regressão DNPM após 1 ano de vida. Diagnóstico com 4 anos, apresentando eczemas em pele, sem dismorfias. Possui irmã afetada que não faz parte desta amostra. V.A.V. G smith-lemli-opitz 1 filha de casal consangüíneo com história de ADNPM e exame dismorfológico característico. Possui irmão afetado que faz parte desta amostra. G.A.V. G smith-lemli-opitz 2 filho de casal consangüíneo com ADNPM e exame dismorfológico característico. Possui irmã afetada que faz parte desta amostra EXAME MOLECULAR O protocolo de PCR estabelecido no laboratório foi aplicado em 34 pacientes do sexo masculino. Em todos os pacientes obteve-se a amplificação por PCR dos produtos polimórficos de repetição da trinca de bases CGG (lócus do FRAXA com 506 e 517 pb), e da trinca de bases CCG (lócus do

71 FRAXE com 419 pb), além da banda controle de 220 pb, menos em um paciente (1) em que foi realizado diagnóstico de Síndrome do X-frágil (Figura 7 e FOTO N). 506/517 pb FRAXA 220 pb 1 c+ c - Figura 7: Fotografia do gel de poliacrilamida a 5%, corado pelo método de prata para a análise dos produtos de PCR e a investigação do lócus FRAXA. Observamos na fotografia, da direita para esquerda: controle negativo (c-) apresentando as bandas de 220 pb e as duas bandas de 506 e 517 pb; controle positivo (c+) amplificando apenas o fragmento de controle interno do experimento; paciente (1) com FRAXA..
 A.A.S. Face longa * Orelhas grandes e")

72 CARACTERÍSTICAS FRAXA PACIENTE (1) A.A.S. Face longa * Orelhas grandes e proeminentes TDAH * Comportamento autista Calosidade em mãos Hipermobilidade articular Palato alto * Macroorquidismo História familiar * FOTO N: Características do paciente com Síndrome do X-frágil. HEREDOGRAMA
Avaliação em DI - Protocolo Clínico
 Avaliação em DI - Protocolo Clínico 1. Avaliação psico-social 2. Avaliação Médica, com ênfase no exame neurológico e genético-clínico. 3. Heredogramas e Detecção de Famílias com múltiplos afetados e consangüinidade.
Avaliação em DI - Protocolo Clínico 1. Avaliação psico-social 2. Avaliação Médica, com ênfase no exame neurológico e genético-clínico. 3. Heredogramas e Detecção de Famílias com múltiplos afetados e consangüinidade.
CONCURSO PÚBLICO ESPECIALISTA DE LABORATÓRIO EDITAL IB ATAD
 CONCURSO PÚBLICO ESPECIALISTA DE LABORATÓRIO EDITAL IB ATAD 24-2012 1ª Prova: Múltipla Escolha Data: 05-09-2012 INSTRUÇÕES PARA FAZER A PROVA: 1) COLOCAR NOME EM TODAS AS PÁGINAS 2) ESCREVER DE FORMA LEGÍVEL
CONCURSO PÚBLICO ESPECIALISTA DE LABORATÓRIO EDITAL IB ATAD 24-2012 1ª Prova: Múltipla Escolha Data: 05-09-2012 INSTRUÇÕES PARA FAZER A PROVA: 1) COLOCAR NOME EM TODAS AS PÁGINAS 2) ESCREVER DE FORMA LEGÍVEL
A criança portadora de deficiência
 A criança portadora de deficiência Unifenas - FCM Pediatria e Puericultura Prof. Dr. Orlando A. Pereira Pessoas Portadoras de Deficiências São pessoas que apresentam significativas diferenças físicas,
A criança portadora de deficiência Unifenas - FCM Pediatria e Puericultura Prof. Dr. Orlando A. Pereira Pessoas Portadoras de Deficiências São pessoas que apresentam significativas diferenças físicas,
Genes e Epilepsia: Epilepsia e Alterações Genéticas
 Faculdade de Medicina de São José do Rio Preto Genes e Epilepsia: Epilepsia e Alterações Genéticas Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo Rodrigo Nunes Cal Introdução Epilepsia
Faculdade de Medicina de São José do Rio Preto Genes e Epilepsia: Epilepsia e Alterações Genéticas Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo Rodrigo Nunes Cal Introdução Epilepsia
Como Conduzir o Recém- Nascido com Malformações. Sessão Clínica da Pediatria Angelina Acosta - FAMEB/UFBA
 Como Conduzir o Recém- Nascido com Malformações Sessão Clínica da Pediatria Angelina Acosta - FAMEB/UFBA Dismorfologia Anomalias Dismórficas Qualquer parte do corpo gravidade variável heterogeneidade etiológica
Como Conduzir o Recém- Nascido com Malformações Sessão Clínica da Pediatria Angelina Acosta - FAMEB/UFBA Dismorfologia Anomalias Dismórficas Qualquer parte do corpo gravidade variável heterogeneidade etiológica
HERANÇA MULTIFATORIAL
 HERANÇA MULTIFATORIAL Resulta de uma combinação de PEQUENAS VARIAÇÕES nos genes que juntas podem produzir ou predispor a um grave defeito, em geral EM CONJUNTO COM FATORES AMBIENTAIS. Tendem a recorrer
HERANÇA MULTIFATORIAL Resulta de uma combinação de PEQUENAS VARIAÇÕES nos genes que juntas podem produzir ou predispor a um grave defeito, em geral EM CONJUNTO COM FATORES AMBIENTAIS. Tendem a recorrer
A A SÍNDROME DO CROMOSSOMO X FRÁGIL Ângela Maria Vianna Morgante IB-USP 1 - INTRODUÇÃO
 A A SÍNDROME DO CROMOSSOMO X FRÁGIL Ângela Maria Vianna Morgante IB-USP 1 - INTRODUÇÃO Já no século XIX se tinha observado que, entre os indivíduos internados em instituições para retardados mentais, a
A A SÍNDROME DO CROMOSSOMO X FRÁGIL Ângela Maria Vianna Morgante IB-USP 1 - INTRODUÇÃO Já no século XIX se tinha observado que, entre os indivíduos internados em instituições para retardados mentais, a
PADRÃO DE HERANÇA LIGADA AO CROMOSSOMO X
 PADRÃO DE HERANÇA LIGADA AO CROMOSSOMO X HOMENS: apresenta um X e um Y XY sexo heterogamético o seus gametas serão metade com cromossomo X e metade com cromossomo Y MULHER: apresenta dois X XX sexo homogamético
PADRÃO DE HERANÇA LIGADA AO CROMOSSOMO X HOMENS: apresenta um X e um Y XY sexo heterogamético o seus gametas serão metade com cromossomo X e metade com cromossomo Y MULHER: apresenta dois X XX sexo homogamético
Biologia. Mutações e Aconselhamento Genético. Professor Enrico Blota.
 Biologia Mutações e Aconselhamento Genético Professor Enrico Blota www.acasadoconcurseiro.com.br Biologia MUTAÇÕES Mutação é uma mudança ou alteração no conteúdo genético de uma célula ou de um ser vivo.
Biologia Mutações e Aconselhamento Genético Professor Enrico Blota www.acasadoconcurseiro.com.br Biologia MUTAÇÕES Mutação é uma mudança ou alteração no conteúdo genético de uma célula ou de um ser vivo.
AS ANOMALIAS CROMOSSÔMICAS EM HUMANOS
 AS ANOMALIAS CROMOSSÔMICAS EM HUMANOS Incidência global de anormalidades cromossômicas em recém-nascidos = 1 em 160 nascimentos. Frequência total de anormalidades cromossômicas em abortos espontâneos =
AS ANOMALIAS CROMOSSÔMICAS EM HUMANOS Incidência global de anormalidades cromossômicas em recém-nascidos = 1 em 160 nascimentos. Frequência total de anormalidades cromossômicas em abortos espontâneos =
O ovo ou zigoto tem um conjunto de cromossomas que provêm dos dois gâmetas, isto é, possui pares de cromossomas homólogos.
 O ovo ou zigoto tem um conjunto de cromossomas que provêm dos dois gâmetas, isto é, possui pares de cromossomas homólogos. Cromossomas homólogos - cromossomas com forma e estrutura idênticas e portadores
O ovo ou zigoto tem um conjunto de cromossomas que provêm dos dois gâmetas, isto é, possui pares de cromossomas homólogos. Cromossomas homólogos - cromossomas com forma e estrutura idênticas e portadores
Microcefalia na atenção básica
 Microcefalia na atenção básica Enfoque da Medicina Fetal Dra. Jamile Simas Abi Saab MICROCEFALIA NA ATENÇÃO BÁSICA Microcefalia: malformação congênita em que o cérebro não se desenvolve de maneira adequada.
Microcefalia na atenção básica Enfoque da Medicina Fetal Dra. Jamile Simas Abi Saab MICROCEFALIA NA ATENÇÃO BÁSICA Microcefalia: malformação congênita em que o cérebro não se desenvolve de maneira adequada.
ESTUDO GENÉTICO-CLÍNICO DE DEFICIENTES MENTAIS INSTITUCIONALIZADOS.
 UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO ESTUDO GENÉTICO-CLÍNICO DE DEFICIENTES MENTAIS INSTITUCIONALIZADOS. Lisandra Mesquita Batista Dissertação de Mestrado apresentada ao Departamento
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO ESTUDO GENÉTICO-CLÍNICO DE DEFICIENTES MENTAIS INSTITUCIONALIZADOS. Lisandra Mesquita Batista Dissertação de Mestrado apresentada ao Departamento
TEA Módulo 1 Aula 5 Genética no Autismo
 TEA Módulo 1 Aula 5 Genética no Autismo Atualmente, a genética é uma das áreas com maior amplitude de aplicação e de pesquisa. As primeiras evidências de que o Autismo pudesse ser resultante de fenômenos
TEA Módulo 1 Aula 5 Genética no Autismo Atualmente, a genética é uma das áreas com maior amplitude de aplicação e de pesquisa. As primeiras evidências de que o Autismo pudesse ser resultante de fenômenos
Causas Microcefalia é o resultado do crescimento abaixo do normal do cérebro da criança ainda no útero ou na infância. A microcefalia pode ser
 Microcefalia Microcefalia Microcefalia é uma condição neurológica rara em que a cabeça e o cérebro da criança são significativamente menores do que os de outras da mesma idade e sexo. A microcefalia normalmente
Microcefalia Microcefalia Microcefalia é uma condição neurológica rara em que a cabeça e o cérebro da criança são significativamente menores do que os de outras da mesma idade e sexo. A microcefalia normalmente
dificuldades de Aprendizagem X distúrbio de Aprendizagem
 Capacitação Multidisciplinar Continuada Como lidar com as dificuldades de Aprendizagem X distúrbio de Aprendizagem O que é aprendizagem Aprendizagem é um processo de mudança de comportamento obtido através
Capacitação Multidisciplinar Continuada Como lidar com as dificuldades de Aprendizagem X distúrbio de Aprendizagem O que é aprendizagem Aprendizagem é um processo de mudança de comportamento obtido através
Objetivos Conceitos Procedimentos e habilidades Atitudes e valores aspectos Comparar cromossomos mitóticos
 Unidade Universitária: CENTRO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE PROGRAMA DE PÓ-GRADUAÇÃO EM DISTÚRBIOS DO DESENVOLVIMENTO Disciplina: Genética dos Distúrbios do Desenvolvimento Código da Disciplina: 900.1235-6
Unidade Universitária: CENTRO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE PROGRAMA DE PÓ-GRADUAÇÃO EM DISTÚRBIOS DO DESENVOLVIMENTO Disciplina: Genética dos Distúrbios do Desenvolvimento Código da Disciplina: 900.1235-6
Aconselhamento Genético e Diagnóstico Pré-natal
 Aconselhamento Genético e Diagnóstico Pré-natal O aconselhamento genético constitui-se de um processo de comunicação que trata dos problemas humanos associados com a ocorrência, ou risco de ocorrência
Aconselhamento Genético e Diagnóstico Pré-natal O aconselhamento genético constitui-se de um processo de comunicação que trata dos problemas humanos associados com a ocorrência, ou risco de ocorrência
Herança multifatorial
 Introdução Herança multifatorial Conceito Fundamentos básicos da herança multifatorial Modelo básico Distribuição normal sino 1 Exemplos de características multifatoriais O modelo de limiar Normais Altura
Introdução Herança multifatorial Conceito Fundamentos básicos da herança multifatorial Modelo básico Distribuição normal sino 1 Exemplos de características multifatoriais O modelo de limiar Normais Altura
A Prevenção do retardo mental na Síndrome do X Frágil
 LOGO A Prevenção do retardo mental na Síndrome do X Frágil Renata Ríspoli Gatti, Msc. Laboratório de Genética Humana Classificação > 200 CGG ~55 200 CGG Afetados Pré mutação 40 ~55 CGG Zona Gray 6 - ~40
LOGO A Prevenção do retardo mental na Síndrome do X Frágil Renata Ríspoli Gatti, Msc. Laboratório de Genética Humana Classificação > 200 CGG ~55 200 CGG Afetados Pré mutação 40 ~55 CGG Zona Gray 6 - ~40
AVALIAÇÃO DO ESTILO PARENTAL DE PAIS DE CRIANÇAS PORTADORAS DE SÍNDROMES GENÉTICAS NA TRÍPLICE FRONTEIRA
 CIÊNCIAS DA SAÚDE AVALIAÇÃO DO ESTILO PARENTAL DE PAIS DE CRIANÇAS PORTADORAS DE SÍNDROMES GENÉTICAS NA TRÍPLICE FRONTEIRA VOLPATO VIEIRA, Marília. Estudante do Curso de Medicina ILACVN UNILA; E-mail:
CIÊNCIAS DA SAÚDE AVALIAÇÃO DO ESTILO PARENTAL DE PAIS DE CRIANÇAS PORTADORAS DE SÍNDROMES GENÉTICAS NA TRÍPLICE FRONTEIRA VOLPATO VIEIRA, Marília. Estudante do Curso de Medicina ILACVN UNILA; E-mail:
CONCEITOS BÁSICOS EM GENÉTICA
 CONCEITOS BÁSICOS EM GENÉTICA HISTÓRICO: Veio ao Serviço de Aconselhamento Genético do Hospital de Clínicas, um casal jovem com o seguinte relato: a) homem fenotipicamente normal, com 35 anos, obeso, pertencente
CONCEITOS BÁSICOS EM GENÉTICA HISTÓRICO: Veio ao Serviço de Aconselhamento Genético do Hospital de Clínicas, um casal jovem com o seguinte relato: a) homem fenotipicamente normal, com 35 anos, obeso, pertencente
Determinação e Diferenciação Sexuais
 Determinação e Diferenciação Sexuais Profa. Dra. Ester Silveira Ramos Faculdade de Medicina de Ribeirão Preto Universidade de São Paulo esramos@rge.fmrp.usp.br Sexo genético Sexo gonadal Genitália interna
Determinação e Diferenciação Sexuais Profa. Dra. Ester Silveira Ramos Faculdade de Medicina de Ribeirão Preto Universidade de São Paulo esramos@rge.fmrp.usp.br Sexo genético Sexo gonadal Genitália interna
Análises moleculares - DNA
 Análises moleculares - DNA Como o mapeamento genético contribui para a genética médica? A caracterização de um gene e suas mutações aumenta a compreensão da doença Aplicações: -Desenvolvimento de diagnóstico
Análises moleculares - DNA Como o mapeamento genético contribui para a genética médica? A caracterização de um gene e suas mutações aumenta a compreensão da doença Aplicações: -Desenvolvimento de diagnóstico
1. Na família abaixo há três indivíduos afetados por uma doença neurológica muito rara.
 1. Na família abaixo há três indivíduos afetados por uma doença neurológica muito rara. I Dê as razões para que essa doença tenha ou não tenha herança: a ligada ao X dominante. Não é esse o padrão de herança,
1. Na família abaixo há três indivíduos afetados por uma doença neurológica muito rara. I Dê as razões para que essa doença tenha ou não tenha herança: a ligada ao X dominante. Não é esse o padrão de herança,
Herança multifatorial
 Introdução Herança multifatorial Conceito Fundamentos básicos da herança multifatorial Modelo básico Distribuição normal sino 1 Exemplos de características multifatoriais O modelo de limiar Normais Altura
Introdução Herança multifatorial Conceito Fundamentos básicos da herança multifatorial Modelo básico Distribuição normal sino 1 Exemplos de características multifatoriais O modelo de limiar Normais Altura
REGISTO NACIONAL DE AN MALIAS CONGÉNITAS O RENAC Resumo
 REGISTO NACIONAL DE AN MALIAS CONGÉNITAS O RENAC 2-21 Resumo Instituto Nacional de Saúde Doutor Ricardo Jorge, IP Lisboa, 28 de novembro de 214 O REGISTO NACIONAL DE ANOMALIAS CONGÉNITAS MATERIAIS E MÉTODOS
REGISTO NACIONAL DE AN MALIAS CONGÉNITAS O RENAC 2-21 Resumo Instituto Nacional de Saúde Doutor Ricardo Jorge, IP Lisboa, 28 de novembro de 214 O REGISTO NACIONAL DE ANOMALIAS CONGÉNITAS MATERIAIS E MÉTODOS
SÍNDROME DE ASPERGER E AUTISMO
 SÍNDROME DE ASPERGER E AUTISMO CASTRO.M.B. 1 ; MARRONI.N.M.O. 2 ; FARIA.M.C.C. 3 ; RESUMO A Síndrome de Asperger é uma desordem pouco comum, ou seja, um grupo de problemas que algumas crianças tem quando
SÍNDROME DE ASPERGER E AUTISMO CASTRO.M.B. 1 ; MARRONI.N.M.O. 2 ; FARIA.M.C.C. 3 ; RESUMO A Síndrome de Asperger é uma desordem pouco comum, ou seja, um grupo de problemas que algumas crianças tem quando
Política Nacional de Atenção Integral às Pessoas com Doenças Raras no SUS. Brasília, 29 de maio de 2014
 Política Nacional de Atenção Integral às Pessoas com Doenças Raras no SUS Brasília, 29 de maio de 2014 Doenças raras OMS: afeta até 65 pessoas/100 mil indivíduos (1,3:2.000). Acometem de 6% a 8% da população.
Política Nacional de Atenção Integral às Pessoas com Doenças Raras no SUS Brasília, 29 de maio de 2014 Doenças raras OMS: afeta até 65 pessoas/100 mil indivíduos (1,3:2.000). Acometem de 6% a 8% da população.
DISTÚRBIOS, TRANSTORNOS, DIFICULDADES E PROBLEMAS DE APRENDIZAGEM
 DISTÚRBIOS, TRANSTORNOS, DIFICULDADES E PROBLEMAS DE APRENDIZAGEM Os termos distúrbios, transtornos, dificuldades e problemas de aprendizagem tem sido utilizados de forma aleatória, tanto na literatura
DISTÚRBIOS, TRANSTORNOS, DIFICULDADES E PROBLEMAS DE APRENDIZAGEM Os termos distúrbios, transtornos, dificuldades e problemas de aprendizagem tem sido utilizados de forma aleatória, tanto na literatura
Glossário. Formas alternativas de um gene no mesmo locus (posição no cromossoma). Uma pessoa herda um alelo do pai e outro alelo da mãe
. Uma pessoa herda um alelo do pai e outro alelo da mãe") Glossário aborto espontâneo aconselhamento genético alelos aneuploidia anomalia cromossómica autossómico recessivo BRCA cariótipo caso índice citogeneticista Perda gestacional de um feto não viável antes
Glossário aborto espontâneo aconselhamento genético alelos aneuploidia anomalia cromossómica autossómico recessivo BRCA cariótipo caso índice citogeneticista Perda gestacional de um feto não viável antes
SERVIÇO PÚBLICO FEDERAL UNIVERSIDADE FEDERAL DE GOIÁS CÃMPUS JATAÍ PLANO DE ENSINO
 PLANO DE ENSINO I. IDENTIFICAÇÃO Unidade Acadêmica: Câmpus Jataí Curso: Biomedicina Disciplina:Genética Humana Matriz: 35P Carga horária semestral: 64h Teórica: 48h Prática: 16h Semestre/ano: 2013/1 Turma/turno:Matutino
PLANO DE ENSINO I. IDENTIFICAÇÃO Unidade Acadêmica: Câmpus Jataí Curso: Biomedicina Disciplina:Genética Humana Matriz: 35P Carga horária semestral: 64h Teórica: 48h Prática: 16h Semestre/ano: 2013/1 Turma/turno:Matutino
CARTILHA DE ORIENTAÇÃO AOS CHEFES
 CARTILHA DE ORIENTAÇÃO AOS CHEFES Visão do trabalhador com deficiência intelectual Instituição beneficente que promove o desenvolvimento e a inserção social de pessoas com deficiência intelectual CARTILHA
CARTILHA DE ORIENTAÇÃO AOS CHEFES Visão do trabalhador com deficiência intelectual Instituição beneficente que promove o desenvolvimento e a inserção social de pessoas com deficiência intelectual CARTILHA
aula 6: quantificação de eventos em saúde
 ACH-1043 Epidemiologia e Microbiologia aula 6: quantificação de eventos em saúde Helene Mariko Ueno papoula@usp.br Como quantificar eventos relacionados à saúde? O que medir? Como medir? Quando medir?
ACH-1043 Epidemiologia e Microbiologia aula 6: quantificação de eventos em saúde Helene Mariko Ueno papoula@usp.br Como quantificar eventos relacionados à saúde? O que medir? Como medir? Quando medir?
ATUAÇÃO DA TERAPIA OCUPACIONAL COM SÍNDROMES GENÉTICAS QUE CAUSAM DEFICIÊNCIA INTELECTUAL
 ATUAÇÃO DA TERAPIA OCUPACIONAL COM SÍNDROMES GENÉTICAS QUE CAUSAM DEFICIÊNCIA INTELECTUAL Tatit, A. L. D.; Luz, M. S.; Zaboroski, A. P.; Freitas, F. P. M.; Oliveira, J. P. Introdução As síndromes genéticas
ATUAÇÃO DA TERAPIA OCUPACIONAL COM SÍNDROMES GENÉTICAS QUE CAUSAM DEFICIÊNCIA INTELECTUAL Tatit, A. L. D.; Luz, M. S.; Zaboroski, A. P.; Freitas, F. P. M.; Oliveira, J. P. Introdução As síndromes genéticas
MEDIDAS DE PREVENÇÃO NA SAÚDE MENTAL. Prof. João Gregório Neto
 MEDIDAS DE PREVENÇÃO NA SAÚDE MENTAL Prof. João Gregório Neto PREVENÇÃO Ato ou efeito de prevenir-se Disposição ou preparo antecipado e preventivo Precaução, cautela Modo de ver antecipado, premeditado
MEDIDAS DE PREVENÇÃO NA SAÚDE MENTAL Prof. João Gregório Neto PREVENÇÃO Ato ou efeito de prevenir-se Disposição ou preparo antecipado e preventivo Precaução, cautela Modo de ver antecipado, premeditado
SÍNDROME DE DOWN: MORBIDADE E MORTALIDADE
 SÍNDROME DE DOWN: MORBIDADE E MORTALIDADE SASAKI, Camila Nathami. SANTOS, João Vitor Brisolla Acadêmicos do curso de Graduação de Fisioterapia da Faculdade de Ciências Sociais e Agrárias de Itapeva SAVIAN,
SÍNDROME DE DOWN: MORBIDADE E MORTALIDADE SASAKI, Camila Nathami. SANTOS, João Vitor Brisolla Acadêmicos do curso de Graduação de Fisioterapia da Faculdade de Ciências Sociais e Agrárias de Itapeva SAVIAN,
Cromossômicas Monogênicas Poligênicas ou Multifatoriais Mitocondriais
 Distúrbios Monogênicos ou MENDELIANOS DISTÚRBIOS MONOGÊNICOS E PRINCIPAIS PADRÕES DE HERANÇA Profa. Dra. Milena Flória-Santos Departamento de Enfermagem Materno-Infantil e Saúde Pública Escola de Enfermagem
Distúrbios Monogênicos ou MENDELIANOS DISTÚRBIOS MONOGÊNICOS E PRINCIPAIS PADRÕES DE HERANÇA Profa. Dra. Milena Flória-Santos Departamento de Enfermagem Materno-Infantil e Saúde Pública Escola de Enfermagem
Espinha Bífida. Dr. Fábio Agertt
 Espinha Bífida Dr. Fábio Agertt Neurônio Motor Superior/Inferior Espinha Bífida Defeito de fechamento do tubo neural; Variedade de apresentações e gravidade; As características podem ser diagnosticadas
Espinha Bífida Dr. Fábio Agertt Neurônio Motor Superior/Inferior Espinha Bífida Defeito de fechamento do tubo neural; Variedade de apresentações e gravidade; As características podem ser diagnosticadas
Cadernos de. Informação. Científica. Ano 8 nº 12 2013. Síndrome do X Frágil
 Cadernos de Informação Científica Ano 8 nº 12 2013 Síndrome do X Frágil C a d e r n o s d e I n f o r m a ç ã o C i e n t í f i c a definição e causas A síndrome do X frágil (SXF), também conhecida como
Cadernos de Informação Científica Ano 8 nº 12 2013 Síndrome do X Frágil C a d e r n o s d e I n f o r m a ç ã o C i e n t í f i c a definição e causas A síndrome do X frágil (SXF), também conhecida como
Dificuldades De Aprendizado. Lancet 2003; 362:
 Dificuldades De Aprendizado Lancet 2003; 362:811-821 Definições Dificuldade de aprendizado e retardo mental são rótulos aplicados a indivíduos que apresentam um teste de QI abaixo de um certo nível (geralmente
Dificuldades De Aprendizado Lancet 2003; 362:811-821 Definições Dificuldade de aprendizado e retardo mental são rótulos aplicados a indivíduos que apresentam um teste de QI abaixo de um certo nível (geralmente
Vigilância e prevenção das Doenças de transmissão vertical 2016/2017
 Vigilância e prevenção das Doenças de transmissão vertical 2016/2017 Principais Doenças de Transmissão Vertical no Brasil Sífilis congênita HIV-AIDS Hepatites B e C Rubéola congênita Toxoplasmose congênita
Vigilância e prevenção das Doenças de transmissão vertical 2016/2017 Principais Doenças de Transmissão Vertical no Brasil Sífilis congênita HIV-AIDS Hepatites B e C Rubéola congênita Toxoplasmose congênita
A análise da evidência para esta diretriz se baseia em dois processos. detalhados de revisão anteriores. O primeiro foi a conferência de consenso dos
 Material Suplementar On-line MÉTODOS E PROCESSO A análise da evidência para esta diretriz se baseia em dois processos detalhados de revisão anteriores. O primeiro foi a conferência de consenso dos National
Material Suplementar On-line MÉTODOS E PROCESSO A análise da evidência para esta diretriz se baseia em dois processos detalhados de revisão anteriores. O primeiro foi a conferência de consenso dos National
Imagem 1 Corpos de Lafora em biópsia axilar corados com Hematoxilina e Eosina (esquerda) e PAS (direita). Fonte: Gökdemir et al, 2012.
 e PAS (direita). Fonte: Gökdemir et al, 2012.") Introdução A doença de Lafora (DL) é a forma mais comum de epilepsia mioclônica progressiva na adolescência. Trata-se de uma doença autossômica recessiva, causada por mutações em genes do metabolismo do
Introdução A doença de Lafora (DL) é a forma mais comum de epilepsia mioclônica progressiva na adolescência. Trata-se de uma doença autossômica recessiva, causada por mutações em genes do metabolismo do
Capítulo 15 Perinatologia PATOLOGIA PERINATAL
 Capítulo 15 Perinatologia PATOLOGIA PERINATAL Tanto as patologias como as anomalias que têm origem no período perinatal estão classificadas no capítulo 15 da CID-9-MC e categorias 760 779. LOCALIZAÇÃO
Capítulo 15 Perinatologia PATOLOGIA PERINATAL Tanto as patologias como as anomalias que têm origem no período perinatal estão classificadas no capítulo 15 da CID-9-MC e categorias 760 779. LOCALIZAÇÃO
Determinantes do processo saúde-doença. Identificação de riscos à saúde. Claudia Witzel
 Determinantes do processo saúde-doença. Identificação de riscos à saúde Claudia Witzel CONCEITOS DE SAÚDE E DOENÇA Saúde pode ser definida como ausência de doença Doença ausência de saúde... Saúde é um
Determinantes do processo saúde-doença. Identificação de riscos à saúde Claudia Witzel CONCEITOS DE SAÚDE E DOENÇA Saúde pode ser definida como ausência de doença Doença ausência de saúde... Saúde é um
Relato de Caso de Síndrome de Smith- Lemli-Optiz em hospital de ensino
 Carneiro, MAD; do Carmo, AV ; Diniz, DS Introdução Faz-se necessário o conhecimento das síndromes genéticas para a prática clínica do médico neurologista. Neste contexto, a Síndrome de Smith-Lemli-Optiz
Carneiro, MAD; do Carmo, AV ; Diniz, DS Introdução Faz-se necessário o conhecimento das síndromes genéticas para a prática clínica do médico neurologista. Neste contexto, a Síndrome de Smith-Lemli-Optiz
Alterações do material genético
 Alterações do material genético Alterações do material genético Agentes internos ou externos causam alterações nos genes ou nos cromossomas MUTAÇÕES (ex: anemia falciforme, trissomia 21) Tecnologia permite
Alterações do material genético Alterações do material genético Agentes internos ou externos causam alterações nos genes ou nos cromossomas MUTAÇÕES (ex: anemia falciforme, trissomia 21) Tecnologia permite
Uso de Medicação Psicotrópica em uma Grande Instituição para Deficientes Mentais - I(1)
") D Deficiência Mental / Déficit Sensorial / Delírio / Depressão / Desenvolvimento / Diabetes Melitus / Diagnóstico / Distúrbio de Leitura / Doença / Doença de Moyamoya / Drogadição DEFICIÊNCIA MENTAL Uso
D Deficiência Mental / Déficit Sensorial / Delírio / Depressão / Desenvolvimento / Diabetes Melitus / Diagnóstico / Distúrbio de Leitura / Doença / Doença de Moyamoya / Drogadição DEFICIÊNCIA MENTAL Uso
TÍTULO: SEXAGEM FETAL:DIAGNÓTICO DO SEXO DO FETO POR REAÇÃO EM CADEIA DE POLIMERASE (PCR)
") TÍTULO: SEXAGEM FETAL:DIAGNÓTICO DO SEXO DO FETO POR REAÇÃO EM CADEIA DE POLIMERASE (PCR) CATEGORIA: EM ANDAMENTO ÁREA: CIÊNCIAS BIOLÓGICAS E SAÚDE SUBÁREA: BIOMEDICINA INSTITUIÇÃO: CENTRO UNIVERSITÁRIO
TÍTULO: SEXAGEM FETAL:DIAGNÓTICO DO SEXO DO FETO POR REAÇÃO EM CADEIA DE POLIMERASE (PCR) CATEGORIA: EM ANDAMENTO ÁREA: CIÊNCIAS BIOLÓGICAS E SAÚDE SUBÁREA: BIOMEDICINA INSTITUIÇÃO: CENTRO UNIVERSITÁRIO
Sobre a Esclerose Tuberosa e o Tumor Cerebral SEGA
 Sobre a Esclerose Tuberosa e o Tumor Cerebral SEGA A Esclerose Tuberosa, também conhecida como Complexo da Esclerose Tuberosa, é uma desordem genética que atinge entre 1 e 2 milhões de pessoas no mundo
Sobre a Esclerose Tuberosa e o Tumor Cerebral SEGA A Esclerose Tuberosa, também conhecida como Complexo da Esclerose Tuberosa, é uma desordem genética que atinge entre 1 e 2 milhões de pessoas no mundo
UNIVERSIDADE FEDERAL DO ESTADO DO RIO DE JANEIRO CENTRO DE CIÊNCIAS BIOLÓGICAS E DA SAÚDE ESCOLA DE MEDICINA E CIRURGIA
 PROGRAMA DE DISCIPLINA CURSO: MEDICINA DEPARTAMENTO: DEPARTAMENTO DE MEDICINA ESPECIALIZADA DISCIPLINA: NEUROLOGIA CARGA HORÁRIA: 120 HORAS CRÉDITOS: 07 CÓDIGO: SME0013 PROFESSOR: REGINA MARIA PAPAIS ALVARENGA
PROGRAMA DE DISCIPLINA CURSO: MEDICINA DEPARTAMENTO: DEPARTAMENTO DE MEDICINA ESPECIALIZADA DISCIPLINA: NEUROLOGIA CARGA HORÁRIA: 120 HORAS CRÉDITOS: 07 CÓDIGO: SME0013 PROFESSOR: REGINA MARIA PAPAIS ALVARENGA
Cromossomos sexuais e suas anomalias
 Cromossomos sexuais e suas anomalias Síndrome de Turner ou Monossomia do cromossomo X A Síndrome de Turner, descrita na década de 40, é característica do sexo feminino e ocorre numa proporção de 1:2500
Cromossomos sexuais e suas anomalias Síndrome de Turner ou Monossomia do cromossomo X A Síndrome de Turner, descrita na década de 40, é característica do sexo feminino e ocorre numa proporção de 1:2500
TRANSTORNO ESPECÍFICO DA APRENDIZAGEM CRITÉRIOS DIAGNÓSTICOS E POSSIBILIDADES DE INTERVENÇÃO
 TRANSTORNO ESPECÍFICO DA APRENDIZAGEM CRITÉRIOS DIAGNÓSTICOS E POSSIBILIDADES DE INTERVENÇÃO Dra Nadia Aparecida Profa. Dra. Nádia Aparecida Doutora em Psicologia e Educação USP, Mestre em Psicologia da
TRANSTORNO ESPECÍFICO DA APRENDIZAGEM CRITÉRIOS DIAGNÓSTICOS E POSSIBILIDADES DE INTERVENÇÃO Dra Nadia Aparecida Profa. Dra. Nádia Aparecida Doutora em Psicologia e Educação USP, Mestre em Psicologia da
HABILITAÇÃO E REABILITAÇÃO DA PESSOA COM DEFICIÊNCIA INTELECTUAL NA PERSPECTIVA DA SAÚDE
 HABILITAÇÃO E REABILITAÇÃO DA PESSOA COM DEFICIÊNCIA INTELECTUAL NA PERSPECTIVA DA SAÚDE AUTORES: Renatha de Carvalho (renathacfisio@gmail.com), Andréa de Jesus Lopes (andrealopesfisio@gmail.com. CER II
HABILITAÇÃO E REABILITAÇÃO DA PESSOA COM DEFICIÊNCIA INTELECTUAL NA PERSPECTIVA DA SAÚDE AUTORES: Renatha de Carvalho (renathacfisio@gmail.com), Andréa de Jesus Lopes (andrealopesfisio@gmail.com. CER II
PERFIL MOTOR DE ESCOLARES SOBREPESOS E OBESOS DE AMBOS OS SEXOS NA FAIXA ETÁRIA DE 9 E 10 ANOS
 PERFIL MOTOR DE ESCOLARES SOBREPESOS E OBESOS DE AMBOS OS SEXOS NA FAIXA ETÁRIA DE 9 E 10 ANOS Liene Mílcia Ap. Josué Orientadora: Prof. Adj. Tamara Goldberg Co-orientador: Prof. Dr. Milton V. do Prado
PERFIL MOTOR DE ESCOLARES SOBREPESOS E OBESOS DE AMBOS OS SEXOS NA FAIXA ETÁRIA DE 9 E 10 ANOS Liene Mílcia Ap. Josué Orientadora: Prof. Adj. Tamara Goldberg Co-orientador: Prof. Dr. Milton V. do Prado
A síndrome do alcoolismo fetal, SAF, é resultado da ingestão de bebida alcoólica pela mãe durante o período de gestação.
 Quando Malcolm nasceu, meu coração se partiu, ela disse. E a culpa, meu Deus, o sentimento de culpa... Quando descobriu que estava grávida, Ellen O Donovan estava perdendo a luta contra o alcoolismo. Meses
Quando Malcolm nasceu, meu coração se partiu, ela disse. E a culpa, meu Deus, o sentimento de culpa... Quando descobriu que estava grávida, Ellen O Donovan estava perdendo a luta contra o alcoolismo. Meses
O X Frágil fica mais forte com você.
 O X Frágil fica mais forte com você. Projeto Eu Digo X SAIBA MAIS SOBRE A SÍNDROME DO X FRÁGIL E NOS AJUDE A CUIDAR MELHOR DE QUEM PRECISA. O X Frágil fica mais forte com você e com toda a sociedade. A
O X Frágil fica mais forte com você. Projeto Eu Digo X SAIBA MAIS SOBRE A SÍNDROME DO X FRÁGIL E NOS AJUDE A CUIDAR MELHOR DE QUEM PRECISA. O X Frágil fica mais forte com você e com toda a sociedade. A
Perfil dos nascidos vivos de mães residentes na área programática 2.2 no Município do Rio de Janeiro
 Perfil dos nascidos vivos de mães residentes na área programática 2.2 no Município do Rio de Janeiro Ana Lucia A. de Toledo Carla R. Fernandes 1 Ana Claudia S. Amaral -NESC/UFRJ-SMS/RJ) Vania da S. Cardoso
Perfil dos nascidos vivos de mães residentes na área programática 2.2 no Município do Rio de Janeiro Ana Lucia A. de Toledo Carla R. Fernandes 1 Ana Claudia S. Amaral -NESC/UFRJ-SMS/RJ) Vania da S. Cardoso
premium Teste de Triagem Pré-natal Não Invasivo em sangue materno
 Software de resultados BGI com marca CE (Conformidade Europeia) para a trissomia 21 Detecta as trissomias fetais dos cromossomos 21, 18 e 13 Informa sobre as trissomias fetais 9, 16 e 22 Informa sobre
Software de resultados BGI com marca CE (Conformidade Europeia) para a trissomia 21 Detecta as trissomias fetais dos cromossomos 21, 18 e 13 Informa sobre as trissomias fetais 9, 16 e 22 Informa sobre
O papel O papel da Genética da Genética na Medicina Introdução 1953
 O papel da Genética na Medicina Profª Ana Luisa Miranda-Vilela Introdução 1953 estrutura em dupla hélice do DNA A partir daí... Aprimoramento das tecnologias de investigação do DNA respostas às questões
O papel da Genética na Medicina Profª Ana Luisa Miranda-Vilela Introdução 1953 estrutura em dupla hélice do DNA A partir daí... Aprimoramento das tecnologias de investigação do DNA respostas às questões
PALAVRAS-CHAVE Morte Fetal. Indicadores de Saúde. Assistência Perinatal. Epidemiologia.
 14. CONEX Apresentação Oral Resumo Expandido - ISSN 2238-9113 1 ISSN 2238-9113 ÁREA TEMÁTICA: (marque uma das opções) ( ) COMUNICAÇÃO ( ) CULTURA ( ) DIREITOS HUMANOS E JUSTIÇA ( ) EDUCAÇÃO ( ) MEIO AMBIENTE
14. CONEX Apresentação Oral Resumo Expandido - ISSN 2238-9113 1 ISSN 2238-9113 ÁREA TEMÁTICA: (marque uma das opções) ( ) COMUNICAÇÃO ( ) CULTURA ( ) DIREITOS HUMANOS E JUSTIÇA ( ) EDUCAÇÃO ( ) MEIO AMBIENTE
UMA SÍNDROME QUE MERECE ATENÇÃO E CARINHO.
 www.eudigox.com.br Expediente Elaboração: Luz Maria Romero e Vanessa Schubert Revisão do conteúdo técnico: Roberto H. Herai Projeto gráfico e diagramação: Agência Fulze Ilustração: Agnes Lina Tsukuda 2
www.eudigox.com.br Expediente Elaboração: Luz Maria Romero e Vanessa Schubert Revisão do conteúdo técnico: Roberto H. Herai Projeto gráfico e diagramação: Agência Fulze Ilustração: Agnes Lina Tsukuda 2
Material e Métodos Resultados e Discussão
 Área: Melhoramento Genético Vegetal TRANSFERIBILIDADE DE PRIMERS MICROSSATÉLITES DE Phaseolus vulgaris PARA Vigna unguiculata Matheus Felipe Nogueira da Silva 1, Leidiane Bezerra Albuquerque 2, Rafaela
Área: Melhoramento Genético Vegetal TRANSFERIBILIDADE DE PRIMERS MICROSSATÉLITES DE Phaseolus vulgaris PARA Vigna unguiculata Matheus Felipe Nogueira da Silva 1, Leidiane Bezerra Albuquerque 2, Rafaela
AVALIAÇÃO DE DESEMPENHO FUNCIONAL DE IDOSOS INSTITUCIONALIZADOS
 AVALIAÇÃO DE DESEMPENHO FUNCIONAL DE IDOSOS INSTITUCIONALIZADOS Cristina Marques de Almeida Holanda¹, Michele Alexandre da Silva². Universidade Federal da Paraíba - UFPB cristinamahd@gmail.com¹, michelebr@live.com
AVALIAÇÃO DE DESEMPENHO FUNCIONAL DE IDOSOS INSTITUCIONALIZADOS Cristina Marques de Almeida Holanda¹, Michele Alexandre da Silva². Universidade Federal da Paraíba - UFPB cristinamahd@gmail.com¹, michelebr@live.com
4. NATALIDADE E MORTALIDADE INFANTIL
 . NATALIDADE E MORTALIDADE INFANTIL .. Introdução A taxa de natalidade e a taxa de mortalidade infantil são indicadores frequentemente utilizados na caracterização da população. O estudo da taxa de natalidade,
. NATALIDADE E MORTALIDADE INFANTIL .. Introdução A taxa de natalidade e a taxa de mortalidade infantil são indicadores frequentemente utilizados na caracterização da população. O estudo da taxa de natalidade,
ULTRASSONOGRAFIA OBSTÉTRICA TRICA E CARDIOPATIAS CONGÊNITAS
 ULTRASSONOGRAFIA OBSTÉTRICA TRICA E CARDIOPATIAS CONGÊNITAS ROSA, Rafael Fabiano Machado; ROSA, Rosana Cardoso Manique; ZEN, Paulo Ricardo Gazzola; KOSHIYAMA, Dayane Bohn; VARELLA- GARCIA, Marileila; PASKULIN,
ULTRASSONOGRAFIA OBSTÉTRICA TRICA E CARDIOPATIAS CONGÊNITAS ROSA, Rafael Fabiano Machado; ROSA, Rosana Cardoso Manique; ZEN, Paulo Ricardo Gazzola; KOSHIYAMA, Dayane Bohn; VARELLA- GARCIA, Marileila; PASKULIN,
Deficiência Intelectual
 Deficiência Intelectual Nicoly Siqueira JOÃO PESSOA-PB COMO ACONTECE Causas de deficiência intelectual no período pré-natal: PERÍODO PRÉ-NATAL BIOMÉDICOS SOCIAIS COMPORTAMENTAIS EDUCACIONAIS Cromossômicas
Deficiência Intelectual Nicoly Siqueira JOÃO PESSOA-PB COMO ACONTECE Causas de deficiência intelectual no período pré-natal: PERÍODO PRÉ-NATAL BIOMÉDICOS SOCIAIS COMPORTAMENTAIS EDUCACIONAIS Cromossômicas
SOBRE MIM... Graduação em nutrição em 2010 pela UNIVERSIDADE ESTADUAL DO CENTRO OESTE UNICENTRO
 SOBRE MIM... Graduação em nutrição em 2010 pela UNIVERSIDADE ESTADUAL DO CENTRO OESTE UNICENTRO Pós graduada em Nutrição Clínica Funcional Unicsul/VP em 2014 Aluna de Mestrado na Universidade Federal de
SOBRE MIM... Graduação em nutrição em 2010 pela UNIVERSIDADE ESTADUAL DO CENTRO OESTE UNICENTRO Pós graduada em Nutrição Clínica Funcional Unicsul/VP em 2014 Aluna de Mestrado na Universidade Federal de
Teste de Triagem Pré-natal Não Invasivo em sangue materno
 Teste de Triagem Pré-natal Não Invasivo em sangue materno Software de resultados BGI com marca CE (Conformidade Europeia) para a trissomia 21 Detecta as trissomias fetais dos cromossomos 21, 18 e 13 Informa
Teste de Triagem Pré-natal Não Invasivo em sangue materno Software de resultados BGI com marca CE (Conformidade Europeia) para a trissomia 21 Detecta as trissomias fetais dos cromossomos 21, 18 e 13 Informa
SDE0183 TEORIA E PRÁTICA DA EDUCAÇÃO FÍSICA ADAPTADA. Aula 9: Atividade Motora Adaptada as PcD s Motoras/Físicas
 SDE0183 TEORIA E PRÁTICA DA EDUCAÇÃO FÍSICA ADAPTADA Aula 9: Atividade Motora Adaptada as PcD s Motoras/Físicas Objetivos desta aula: 1. Conhecer e diferenciar as deficiências motoras e físicas, suas causas
SDE0183 TEORIA E PRÁTICA DA EDUCAÇÃO FÍSICA ADAPTADA Aula 9: Atividade Motora Adaptada as PcD s Motoras/Físicas Objetivos desta aula: 1. Conhecer e diferenciar as deficiências motoras e físicas, suas causas
Zika virus: um novo teratógeno ZIKV microcefalia
 A história natural da epidemia por vírus Zika em Tangará da Serra incidência na população geral, nas gestantes, anomalias congênitas em recém-nascidos e consequências para o desenvolvimento infantil Lavínia
A história natural da epidemia por vírus Zika em Tangará da Serra incidência na população geral, nas gestantes, anomalias congênitas em recém-nascidos e consequências para o desenvolvimento infantil Lavínia
Campus de Botucatu PLANO DE ENSINO ( X ) OBRIGATÓRIA ( ) OPTATIVA. DOCENTE RESPONSÁVEL : Prof. Dr. Marcelo Razera Baruffi
 OBRIGATÓRIA ( ) OPTATIVA. DOCENTE RESPONSÁVEL : Prof. Dr. Marcelo Razera Baruffi") PLANO DE ENSINO I IDENTIFICAÇÃO CURSO: Nutrição DISCIPLINA: Genética e Evolução ( X ) OBRIGATÓRIA ( ) OPTATIVA DEPARTAMENTO: Genética DOCENTE RESPONSÁVEL : Prof. Dr. Marcelo Razera Baruffi Semestre Letivo:
PLANO DE ENSINO I IDENTIFICAÇÃO CURSO: Nutrição DISCIPLINA: Genética e Evolução ( X ) OBRIGATÓRIA ( ) OPTATIVA DEPARTAMENTO: Genética DOCENTE RESPONSÁVEL : Prof. Dr. Marcelo Razera Baruffi Semestre Letivo:
DOENÇAS GENÉTICAS. Classificação das Doenças Genéticas. Distúrbios Monogênicos (Mendeliano)
") DOENÇAS GENÉTICAS Classificação das Doenças Genéticas Distúrbios cromossômicos Distúrbios Monogênicos (Mendeliano) Distúrbios Multifatoriais Distúrbios Oligogênicos e poligênicos Distúrbios Mitocôndriais
DOENÇAS GENÉTICAS Classificação das Doenças Genéticas Distúrbios cromossômicos Distúrbios Monogênicos (Mendeliano) Distúrbios Multifatoriais Distúrbios Oligogênicos e poligênicos Distúrbios Mitocôndriais
SÍNDROME DE DOWN E DIAGNÓSTICO PRÉ-NATAL: PERSPECTIVAS DAS GRÁVIDAS EM RISCO E DAS MÃES DE PORTADORES DE TRISSOMIA 21
 INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR UNIVERSIDADE DO PORTO MESTRADO EM MEDICINA LEGAL SÍNDROME DE DOWN E DIAGNÓSTICO PRÉ-NATAL: PERSPECTIVAS DAS GRÁVIDAS EM RISCO E DAS MÃES DE PORTADORES DE TRISSOMIA
INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR UNIVERSIDADE DO PORTO MESTRADO EM MEDICINA LEGAL SÍNDROME DE DOWN E DIAGNÓSTICO PRÉ-NATAL: PERSPECTIVAS DAS GRÁVIDAS EM RISCO E DAS MÃES DE PORTADORES DE TRISSOMIA
Boletim semanal #2 Resposta da Representação da OPAS/OMS no Brasil para a epidemia do vírus da zika e suas consequências
 Boletim semanal #2 Resposta da Representação da OPAS/OMS no Brasil para a epidemia do vírus da zika e suas consequências 11 de março de 2016 Reunião do Comitê de Emergências discutiu zika, microcefalia
Boletim semanal #2 Resposta da Representação da OPAS/OMS no Brasil para a epidemia do vírus da zika e suas consequências 11 de março de 2016 Reunião do Comitê de Emergências discutiu zika, microcefalia
O trabalho do psicopedagogo domiciliar para a reabilitação cognitiva e neuroaprendizagem de pessoas com deficiências severas
 O trabalho do psicopedagogo domiciliar para a reabilitação cognitiva e neuroaprendizagem de pessoas com deficiências severas Thaianny Cristine Salles da Silva Psicopedagoga, Neuropsicopedagoga, Especialista
O trabalho do psicopedagogo domiciliar para a reabilitação cognitiva e neuroaprendizagem de pessoas com deficiências severas Thaianny Cristine Salles da Silva Psicopedagoga, Neuropsicopedagoga, Especialista
13. CONEX Apresentação Oral Resumo Expandido 1. Perfil epidemiológico da sífilis gestacional em residentes de Ponta Grossa, 2010 a 2014
 13. CONEX Apresentação Oral Resumo Expandido 1 ISSN 2238-9113 ÁREA TEMÁTICA: ( ) COMUNICAÇÃO ( ) CULTURA ( ) DIREITOS HUMANOS E JUSTIÇA ( ) EDUCAÇÃO ( ) MEIO AMBIENTE (X) SAÚDE ( ) TRABALHO ( ) TECNOLOGIA
13. CONEX Apresentação Oral Resumo Expandido 1 ISSN 2238-9113 ÁREA TEMÁTICA: ( ) COMUNICAÇÃO ( ) CULTURA ( ) DIREITOS HUMANOS E JUSTIÇA ( ) EDUCAÇÃO ( ) MEIO AMBIENTE (X) SAÚDE ( ) TRABALHO ( ) TECNOLOGIA
Técnicas utilizadas para estudo citogenético clínico. Prof. Dr. Bruno Lazzari de Lima
 Técnicas utilizadas para estudo citogenético clínico Prof. Dr. Bruno Lazzari de Lima Citogenética Clínica Estudo do cromossomo aplicado à prática da genética médica. 40 anos atrás Distúrbios cromossômicos
Técnicas utilizadas para estudo citogenético clínico Prof. Dr. Bruno Lazzari de Lima Citogenética Clínica Estudo do cromossomo aplicado à prática da genética médica. 40 anos atrás Distúrbios cromossômicos
APRENDER A SUPERAR. Conhecer o que afeta o seu aluno é o primeiro passo para criar estratégias que garantam a aprendizagem
 APRENDER A SUPERAR Conhecer o que afeta o seu aluno é o primeiro passo para criar estratégias que garantam a aprendizagem O QUE É DEFICIÊNCIA? Deficiência é um desenvolvimento insuficiente, em termos globais
APRENDER A SUPERAR Conhecer o que afeta o seu aluno é o primeiro passo para criar estratégias que garantam a aprendizagem O QUE É DEFICIÊNCIA? Deficiência é um desenvolvimento insuficiente, em termos globais
EPILEPSIA PÓS- TRAUMÁTICA
 EPILEPSIA PÓS- TRAUMÁTICA 4º CONGRESSO NACIONAL DE MEDICINA LEGAL Marcos Barbosa Serviço de Neurocirurgia Hospitais da Universidade de Coimbra Coimbra, Portugal Covilhã, Novembro 2005 Epilepsia é uma perturbação
EPILEPSIA PÓS- TRAUMÁTICA 4º CONGRESSO NACIONAL DE MEDICINA LEGAL Marcos Barbosa Serviço de Neurocirurgia Hospitais da Universidade de Coimbra Coimbra, Portugal Covilhã, Novembro 2005 Epilepsia é uma perturbação
Revisão geral 8º ANO.
 Revisão geral 8º ANO. Cromossomos e Determinação do sexo biológico 46 Cromossomos (Total) 44 Cromossomos Autossomos 2 Cromossomos Sexuais Cariótipo e Cariograma XX (Feminino) XY (Masculino) Genes Alelos
Revisão geral 8º ANO. Cromossomos e Determinação do sexo biológico 46 Cromossomos (Total) 44 Cromossomos Autossomos 2 Cromossomos Sexuais Cariótipo e Cariograma XX (Feminino) XY (Masculino) Genes Alelos
Paixão Schlatter, João Carlos Prolla, Patricia Ashton Prolla
 Implementação de protocolo de rastreamento imunohistoquímico de tumores para identificação de predisposição hereditária ao câncer no Sistema Único de Saúde Patrícia Koehler Santos, Silvia Liliana Cossio,
Implementação de protocolo de rastreamento imunohistoquímico de tumores para identificação de predisposição hereditária ao câncer no Sistema Único de Saúde Patrícia Koehler Santos, Silvia Liliana Cossio,
objetivos Ações preventivas AULA
 Ações preventivas 81 11 AULA objetivos Esperamos que, após o estudo do conteúdo desta aula, você seja capaz de: Entender o significado das ações preventivas na vida das pessoas. Compreender que as ações
Ações preventivas 81 11 AULA objetivos Esperamos que, após o estudo do conteúdo desta aula, você seja capaz de: Entender o significado das ações preventivas na vida das pessoas. Compreender que as ações
processos normais relacionados à aquisição e desenvolvimento da audição, voz e fala das crianças.
 Saúde coletiva para a infância Ciclos da Vida Profa. Me. Adriana de Medeiros Melo Membro do Departamento de Saúde Coletiva da Sociedade Brasileira de Fonoaudiologia. Quais são as principais ações da área
Saúde coletiva para a infância Ciclos da Vida Profa. Me. Adriana de Medeiros Melo Membro do Departamento de Saúde Coletiva da Sociedade Brasileira de Fonoaudiologia. Quais são as principais ações da área
Genética Clínica História e Exame Físico
 7 o Congresso Nacional de Pediatria Região Norte - Manaus Genética Clínica História e Exame Físico Prof a Dr a Ana Maria Martins UNIFESP-EPM CONCEITOS Ana Maria Martins UNIFESP -EPM DESVIO FENOTÍPICO:
7 o Congresso Nacional de Pediatria Região Norte - Manaus Genética Clínica História e Exame Físico Prof a Dr a Ana Maria Martins UNIFESP-EPM CONCEITOS Ana Maria Martins UNIFESP -EPM DESVIO FENOTÍPICO:
HEPATITES VIRAIS SEXUALMENTE TRANSMISSÍVEIS EM IDOSOS: BRASIL, NORDESTE E PARAÍBA
 HEPATITES VIRAIS SEXUALMENTE TRANSMISSÍVEIS EM IDOSOS: BRASIL, NORDESTE E PARAÍBA Larissa Ferreira de Araújo Paz (1); Larissa dos Santos Sousa (1) Polyana Cândido de Andrade (2); Gilson Vasco da Silva
HEPATITES VIRAIS SEXUALMENTE TRANSMISSÍVEIS EM IDOSOS: BRASIL, NORDESTE E PARAÍBA Larissa Ferreira de Araújo Paz (1); Larissa dos Santos Sousa (1) Polyana Cândido de Andrade (2); Gilson Vasco da Silva
SÍNDROME DE DOWN. PALAVRAS-CHAVE: Mongolismo; Trissomia do 21; Trissomia do cromossomo 21
 SÍNDROME DE DOWN Isabella garcia vialli 1, rafaella costa olschowsky, jennifer lis quiroga madeira machado, márcia regina terra 2, beatriz machado 2. RESUMO A síndrome de Down é uma condição genética caracterizada
SÍNDROME DE DOWN Isabella garcia vialli 1, rafaella costa olschowsky, jennifer lis quiroga madeira machado, márcia regina terra 2, beatriz machado 2. RESUMO A síndrome de Down é uma condição genética caracterizada
3º - A pessoa com Transtorno do Espectro Autista é considerada pessoa com deficiência, para todos os efeitos legais.
 PROJETO DE LEI Nº /2017 INSTITUI A POLÍTICA MUNICIPAL DOS DIREITOS DA PESSOA COM TRANSTORNO DO ESPECTRO AUTISTA. A Prefeita Municipal de Mossoró, Estado do Rio Grande do Norte, no uso de suas atribuições
PROJETO DE LEI Nº /2017 INSTITUI A POLÍTICA MUNICIPAL DOS DIREITOS DA PESSOA COM TRANSTORNO DO ESPECTRO AUTISTA. A Prefeita Municipal de Mossoró, Estado do Rio Grande do Norte, no uso de suas atribuições
Introdução à Bioquímica
 Introdução à Bioquímica Nucleotídeos e Ácidos Nucléicos Dra. Fernanda Canduri Laboratório de Sistemas BioMoleculares. Departamento de Física.. UNESP São José do Rio Preto - SP. Genoma! O genoma de um organismo
Introdução à Bioquímica Nucleotídeos e Ácidos Nucléicos Dra. Fernanda Canduri Laboratório de Sistemas BioMoleculares. Departamento de Física.. UNESP São José do Rio Preto - SP. Genoma! O genoma de um organismo
Rudimar Riesgo MD, PhD Neuropediatra, Eletrencefalografista Pediátrico Professor de Medicina - UFRGS
 Rudimar Riesgo MD, PhD Neuropediatra, Eletrencefalografista Pediátrico Professor de Medicina - UFRGS TUDO COMEÇOU... MCH - 1997/1998 DESAFIOS CLÍNICOS InTechOpen 10 million visits 27 thousand chapters
Rudimar Riesgo MD, PhD Neuropediatra, Eletrencefalografista Pediátrico Professor de Medicina - UFRGS TUDO COMEÇOU... MCH - 1997/1998 DESAFIOS CLÍNICOS InTechOpen 10 million visits 27 thousand chapters
NOTA TECNICA SAÚDE-N. 25-2015
 NOTA TECNICA SAÚDE-N. 25-2015 Brasília, 30 de novembro de 2015. Área: Área Técnica em Saúde Título: Surto de Microcefalia- o que você precisa saber. Fonte: Dab/MS/SAS 1- Cenário atual O Ministério da Saúde
NOTA TECNICA SAÚDE-N. 25-2015 Brasília, 30 de novembro de 2015. Área: Área Técnica em Saúde Título: Surto de Microcefalia- o que você precisa saber. Fonte: Dab/MS/SAS 1- Cenário atual O Ministério da Saúde