Comparação de sequências
|
|
|
- Gabriel Henrique Alcaide
- 5 Há anos
- Visualizações:
Transcrição

1 Comparação de sequências João Carlos Setubal /28/2018 J. C. Setubal 1
2 Motivação

3 Ciccarelli et al, Science, 2006
4 Como foi possível criar tal árvore? Determinar genes que são compartilhados por todos os seres vivos O que é compartilhar? Gene x 1 em organismo A tem função α Gene x 2 em organismo B é homólogo e tem a mesma função α Então x 1 e x 2 são o mesmo gene x e portanto ele é compartilhado por A e B Quais genes são esses?
5
6 Questões Onde podemos achar as sequências dos genes? Como determinar compartilhamento? Como preparar esses dados para construir uma árvore? Como construir uma árvore? Como saber se ela esta correta?

7 Fig. 1. Overview of the procedure. F D Ciccarelli et al. Science 2006;311: Published by AAAS
8 Respostas Onde podemos achar as sequências dos genes? Bancos de dados públicos (NCBI) BLAST Como determinar compartilhamento? Através de comparação de sequências Como preparar esses dados para construir uma árvore? Alinhamento múltiplo concatenado Como construir uma árvore? Métodos de reconstrução filogenética Como saber se ela esta correta? Inferência é um termo melhor do que construção Argumentos probabilísticos Transferência Horizontal (Lateral) de Genes
9 Por que comparar sequências? Achar similaridades Dadas 2 sequências, quão parecidas elas são? DNA e proteína Buscas em banco de dados Achar quais sequências do banco são parecidas com minha sequência-consulta Consulta (query) é tipicamente uma sequência nova google
10 Motivação (cont.) Construir famílias de proteínas Saber quais organismos tem membros da família Determinar uma assinatura para a família Construir filogenias Entender a história evolutiva de genes e organismos
11 Premissas Em geral buscamos sequências aparentadas Sequências aparentadas são similares aparentadas = homólogas Descendem de um mesmo ancestral Descendentes sofreram mutações ao longo do tempo

12 Artigo publicado em agosto/2018
13 Métodos The dataset consists of 102 species and 29 universally distributed, protein-coding genes Proteomes were downloaded from GenBank and putative orthologues were identified using BLAST. The top hits were compiled and aligned into gene-specific files in MUSCLE
14 Alinhamento de DNA GTGGTGGCCTACGAAGGT GTAGTGCCTTCGAAGGGT
15 Como avaliar um alinhamento? Sistema de pontuação Match: +1 Mismatch: 1
16 Pontuação do alinhamento GTGGTGGCCTACGAAGGT GTAGTGCCTTCGAAGGGT = 4
17 É possível melhorar o alinhamento? Sim Pela introdução de espaços
18 Alinhamento com espaços GTGGTGGCCTACGAA-GGT GTAGTG-CCTTCGAAGGGT
19 Sistema de pontuação com espaços Match: +1 Mismatch: 1 Espaço: 2 (Buraco: sequência de espaços) Em inglês: gaps
20 Pontuação do alinhamento GTGGTGGCCTACGAA-GGT GTAGTG-CCTTCGAAGGGT = 9
21 Justificativa para o sistema de pontuação Matches tem que ser recompensados (> 0) Mismatches e espaços tem que ser penalizados (< 0) Mismatches representam substituições Mutações (ocorrem com frequência) Podem não trazer letalidade Espaços representam inserções ou remoções Mais prováveis de causarem letalidade Alteram quadro de leitura Ocorrem com muito menos frequência
22 Alinhamentos ótimos São os alinhamentos de pontuação máxima similaridade entre duas sequências É o valor da pontuação do alinhamento ótimo No exemplo anterior Similaridade = 9
23 Comparação de sequências de aminoácidos
24 Pontuação de alinhamento de proteínas % de identidade é uma medida simples mas válida de similaridade de sequências de proteínas
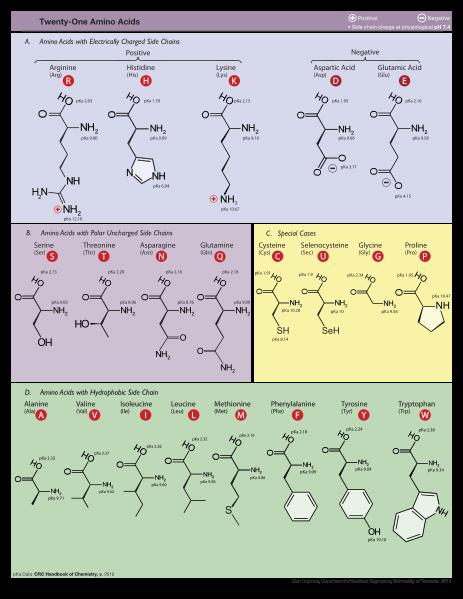
25 Aminoácidos se dividem em famílias Hidrofóbicos Ala, Val, Phe, Pro, Met, Ile, Leu Com carga Asp, Glu, Lys, Arg Polares Gly Ser, Thr, Tyr, His, Cys, Asn, Gln Trp
26
27 Mutações e proteínas Substituições que não alteram a estrutura da proteína tendem a ser preservadas durante a evolução A troca de um aminoácido de uma família por outro da mesma família em geral cai nessa categoria (Indels podem ter consequências mais drásticas) Então: como avaliar mismatches?
28 Matriz de substituição de amino ácidos BLOSUM62 Fonte: NCBI
29 Pontuação leva em conta a matriz Match: blosum62(i,i) sempre positivo Mismatch: blosum62(i,j) positivo, nulo, negativo Espaço: 2
30 Como obter alinhamentos ótimos? Precisamos de um algoritmo Algoritmos são diferentes de programas Algoritmo é um método abstrato Programa é uma encarnação física (numa linguagem de programação) de um algoritmo Um programa pode ser executado num computador Dizemos que um certo algoritmo foi implementado em tal ou qual linguagem Quem escrever algoritimo na prova perde 3 pontos!
31 Algoritmo para achar alinhamentos ótimos de DNA Desenvolvido com a técnica de Programação dinâmica Técnica desenvolvida na década de 1950 por Richard Bellman Não é programação e não é dinâmica!
32 Programação Dinâmica PD se usa para problemas que tem uma estrutura de subproblemas Num alinhamento com sequências s e t um subproblema é qualquer alinhamento entre s e t tal que s = um prefixo de s e t = um prefixo de t Um prefixo 8/28/2018 J. C. Setubal 32
33 Ideia básica da PD Achar soluções de subproblemas e armazená-las numa tabela (matriz) Para achar a solução ótima: Ir achando as soluções na direção dos subproblemas menores para os maiores Último elemento da tabela a ser preenchido contém a solução do problema completo
34 j i t G A T C 0 s 1 G 2 T 3 C
35 Preenchimento da tabela Este processo precisa começar com o menor subproblema possível. Qual seria? Quando pelo menos uma das sequências é vazia Inicialização: Alinhar s com cadeia vazia e alinhar t com cadeia vazia
36 j i t G A T C 0 s G -2 2 T -4 3 C -6
37 Continuação Alinhar caracter X com caracter Y 3 possibilidades X com Y Aplicar pontuação respectiva, dependendo se for DNA ou proteína X com espaço Cobrar -2 Y com espaço Cobrar -2
38 Preenchimento de (1,1) Significa determinar qual é o melhor alinhamento dos prefixos de s e t com apenas um caracter cada um Alternativas G- -G G -G G- G Todos eles usam valores determinados na inicialização
39 j i t G A T C 0 s G T -4 3 C -6
40 j i t G A T C 0 s G T C
41 Exercícios Inventar duas sequências de DNA curtas e rodar (na mão) o algoritmo de PD Para se auto-corrigir: Sequence-Alignment-using-Dynamic-Programming-A (busque dna sequence alignment code project) Demo parece segura Exige registro Tem código fonte
42 Complexidade computacional de PD Queremos saber quanto tempo gasta o algoritmo Mas algoritmos são abstratos, não estão associados a computadores físicos; o que significa tempo? Então contamos número de operações elementares Operações aritméticas Uso de posições da memória Etc Porém queremos apenas algo aproximado Uma ordem de grandeza
43 Complexidade computacional Queremos expressar o número de operações como uma função matemática do tamanho das entradas Por exemplo n (linear) n 2 (quadrático) n log n Vamos desprezar constantes (n e 30n dão na mesma) Só nos interessa o termo de maior grau (desprezar os demais) 7n n log n vira apenas n 2 Vamos usar a notação O(f(n)) para denotar tudo isso
44 Complexidade computacional de PD A matriz tem tamanho n+1 por m+1 Todos os elementos da matriz precisam ser preenchidos Supondo tempo constante para o preenchimento n+1 m+1 = nm + n + m + 1 O(nm) Se n m, O(n 2 ) Quadrático Memória: quadrático também
45
46 Penalização de espaços pode ser mais sofisticada GTGGTGGCCTACGAAGGT GTGGTCGC---CGAAGGT GTGGTGGCC-ACGAAGGT GT-GTCGCCTACGA-GGT No sistema de pontuação apresentado, k espaços consecutivos (um buraco ou gap) custam o mesmo que k espaços separados Seria melhor distinguir os dois casos
47 Penalização de espaços feita por uma função matemática k = número de espaços p(k) = a + bk p(k) é subtraído do score a = custo para abrir um buraco b = custo para continuar um buraco Por exemplo: p(k) = 2 + k 5 espaços consecutivos custam 7 5 espaços separados custam 15 Compare com a função implícita do sistema simples: p(k) = 2k
48 Algoritmo de PD com penalização de buracos por função afim O algoritmo é mais complexo São necessárias 3 tabelas ao invés de 1 Mas a complexidade permanece a mesma (quadrática) Algoritmo de Smith-Waterman
49 Smith-Waterman
50 Se a função for genérica [ex. p(k) = a + b log k)], então a complexidade passa para O(n 3 ) Algoritmo de Needleman-Wunsch
51 Queremos descobrir sequências aparentadas Aparentadas = ancestral comum = homólogas Alinhamentos biologicamente relevantes Nota máxima por si só não nos informa sobre parentesco Alinhamentos de nota máxima não necessariamente correspondem a alinhamentos biologicamente relevantes Como fazer?
52 Bancos de sequências Situação típica Tenho uma sequência consulta Quero saber se existem sequências já publicadas que são parentes dela Tenho que fazer uma busca em bancos de sequências
53 Bancos de sequências Resultado do sequenciamento em geral é publicado bancos de dados de sequências Na verdade catálogos Mais importante: GenBank Mantido pelo National Center for Biotechnological Information NCBI
54

55 UniProt

56 Estatística de alinhamentos Com um banco, temos uma população de sequências Com essa população, posso criar uma teoria estatística que vai me permitir separar os alinhamentos estatisticamente significativos daqueles obtidos por mero acaso Diremos que os alinhamentos estatisticamente significativos são biologicamente relevantes A significância estatística precisa ser quantificada: e-value
57 E-value Teoria de Karlin e Altschul Calcula o e-value (expect value) de um alinhamento E = Kmne λs m e n são os tamanhos das sequências S é a pontuação K e λ são parâmetros Um banco de sequências pode ser tratado como uma longa sequência de tamanho n A fórmula dá o número de alinhamentos que se esperaria obter com pontuação pelo menos S ao acaso
58 e-value Não é uma probabilidade Pode resultar maior do que 1 Mas em geral os alinhamentos biologicamente relevantes tem e-value < 10 5 Para valores assim ou menores, o e-value se comporta como uma probabilidade p-values e e-values P = 1 e E
59 E-value depende do tamanho do banco Não se pode comparar diretamente e-values obtidos de consultas a bancos diferentes Mas existe uma fórmula de conversão Dado o e-value contra banco X, é possível saber qual seria o e-value contra banco Y Essa mesma fórmula pode ser usada para dar o e- value para comparação de apenas duas sequências entre si (supondo que Y seja genbank)

60
61 Programação dinâmica é cara Especialmente quando Comparação contra muitas sequências Buscas em banco de dados Comparação de muitas sequências entre si Todas contra todas Alternativa: BLAST Basic Local Alignment Search Tool
62 BLAST Altschul et al., 1990, 1997 Cerca de 75 mil citações (10/2014) Programa mais citado em ciência (mas não são os papers mais citados; o mais citado tem 305 mil citações) Heurística Não tem garantia de que sempre consegue achar os alinhamentos de pontuação máxima Sacrifica garantia de otimalidade por velocidade Mas na vasta maioria das vezes tais alinhamentos são de fato encontrados Reporta e-values (É possível fazer cálculo de e-values com PD)

63 BLAST Acha alinhamentos locais global
64 É útil pensar na matriz de PD como se fosse um dotplot j i t G A T C 0 s G T C
65 Um alinhamento local é um trecho de células consecutivas num dotplot
66 Pode haver vários alinhamentos locais
67 Alinhamento global
68 Exercício Como modificar o algoritmo de PD para que ele encontre o melhor alinhamento local? Dica: comece definindo as características do melhor alinhamento local
69 Alinhamento local com PD Um alinhamento global pode ter nota negativa um alinhamento local nunca pode ter nota negativa Pois o alinhamento entre sequências vazias tem nota zero bons alinhamentos locais são trechos com notas positivas na matriz de PD
70 Implementação 1. Inicialização da coluna zero e da linha zero com zeros 2. Ao preencher um elemento da matriz, fazer a operação de máximo, mas nunca deixar que o valor escolhido seja negativo Valor max (M[i-1,j], M[i, j-1], M[i-1,j-1], 0) 3. Ao final: procurar o elemento da matriz de valor máximo 4. Para recuperar o alinhamento: percorrer de trás para diante até chegar em zero
71 De onde vem a eficiência de BLAST? BLAST busca trechos parecidos (palavras ou words) entre as sequências = alinhamentossemente Para nt, esses alinhamentos tem que ser exatos Para aa, esses alinhamentos tem que ter nota positiva Estende esses alinhamentos-semente
72 Crédito: I. Lobo
73 Exemplo de alinhamento ótimo que BLAST não encontraria Suponha nt e tamanho mínimo de palavras = 4 GTG-TGGCCTA-GAAGCT GTGGTCG-CTACGACG-T 16 posições, 12 identidades (75%)
74 Características de BLAST Tamanho default das palavras DNA: 11 nt Proteínas: 3 aa Reporta bit score, raw score, e-value, identidades, positivos, buracos

75
76

77 Buscando no GenBank

78 Lista de hits
79 Sabores de BLAST Subject nucleotídeos aminoácidos Query nucleotídeos BLASTN TBLASTX BLASTX aminoácidos TBLASTN BLASTP Também: megablast, psi-blast, phi-blast, delta-blast, etc JC Setubal 79
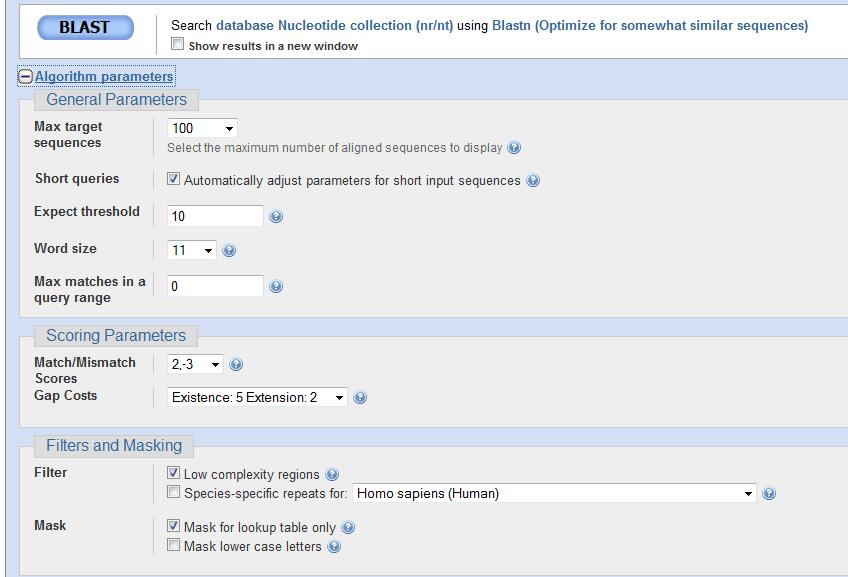
80 Parâmetros de BLASTn
81 Regiões de baixa complexidade Sequências com elementos repetitivos e que aparecem com frequência Exemplo em DNA AAAAAAA Exemplo em proteína AGNLLGRNVVVVGAG Uso do filtro é default Pode excluir alinhamentos relevantes
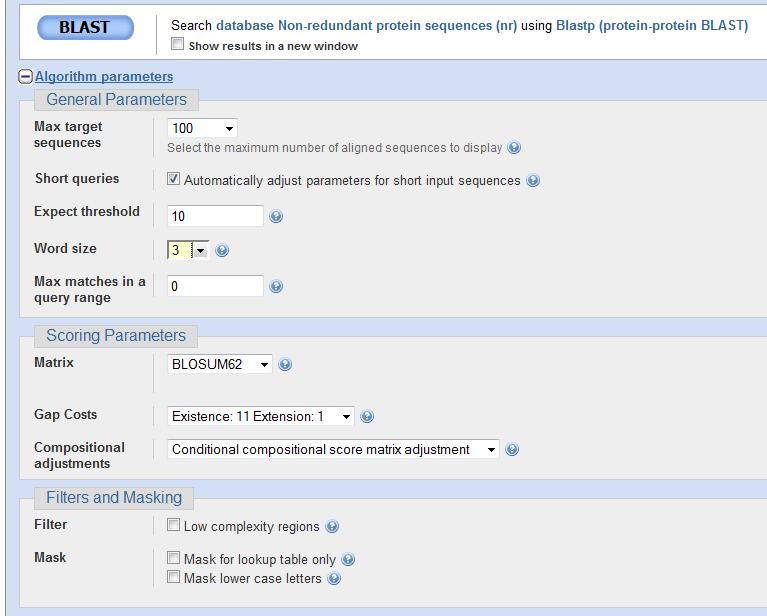
82 Parâmetros de BLASTp
83 Posso acreditar nos resultados do BLAST? 10-5 Falsos negativos Falsos positivos Não significante significante 1) Nem todos os alinhamentos estatisticamente significativos são biologicamente relevantes 2) Nem todos os alinhamentos que não são estatisticamente significantes não são relevantes

84 Exemplo do caso (1) Duas proteínas podem compartilhar um domínio e não serem relacionadas falsos positivos de BLAST Acontece quando as proteínas tem múltiplos domínios Pyruvate kinase

85 Referências Ian Korf, Mark Yandell, Joseph Bedell Artigo por Ingrid Lobo (Write Science Right) 2008 Nature Education

86
87 Novos programas Usearch [Edgar 2010] Até 400x mais rápido do que BLAST Com algum sacrifício de precisão Pauda [Huson e Xie, 2014] Blastx dos pobres x mais rápido do que blastx! Com mais sacrifício de precisão Diamond [Buchfink, Xie, Huson 2014] x mais rápido do que blastx! Sem sacrifício de precisão!
88 Alinhamento múltiplo

89 Alinhamento múltiplo Para construir filogenias é necessário criar AMs JC Setubal 89
90 Avaliação de AMs por soma de pares Numa coluna, podemos separar todos os pares de aminoácidos 1 com 2, 1 com 3, 1 com 4, etc 2 com 3, 2 com 4, etc A cada par corresponde uma nota na matriz BLOSUM62 A soma de todas as notas dos pares dá a nota da coluna A soma das notas das colunas dá a nota do alinhamento
91 Coluna de um alinhamento L I V V I BLOSUM62 L/L: 4 L/I: 2 L/V: 1 I/I: 4 I/V: 3 V/V: 4 L I V V I L I V V 3 3 I 26 Nota da coluna
 indicates positions which have a single, fully conserved residue A : (colon) indicates conservation between groups of strongly similar properties - scoring > 0.")
92 What do the consensus symbols mean in the alignment? An * (asterisk) indicates positions which have a single, fully conserved residue A : (colon) indicates conservation between groups of strongly similar properties - scoring > 0.5 in the Gonnet PAM 250 matrix A. (period) indicates conservation between groups of weakly similar properties - scoring =< 0.5 in the Gonnet PAM 250 matrix
93 Não existe padrão universalmente aceito para avaliar AMs Ou seja, não existe o equivalente de e-values em BLAST Diferentes programas produzem diferentes notas

94 Multiple Sequence Alignment 8/28/2018 J. C. Setubal 94
95 Sequências de entrada Dois conceitos importantes Homologia Família 8/28/2018 J. C. Setubal 95
96 Homologia Dois genes que tem um mesmo ancestral são homológos Freq. usado erroneamente com o sentido de similar Similaridade não implica necessariamente em homologia Asas: morcêgo e insetos (convergência) Às vezes a similaridade é (ou parece) baixa mas mesmo assim existe homologia Barbatana de baleia e braços em humanos Dois tipos de homologia Ortologia e paralogia

97 Ortólogos especiação 28 August 2018 JC Setubal 97
98 parálogos Figure by C. Lasher 28 August 2018 JC Setubal 98
99 In-parálogos Figure by C. Lasher 28 August 2018 JC Setubal 99
100 Homologia e função Seria bom se proteína homólogas tivessem mesma função Geralmente é o caso; mas nem sempre Parálogos estão mais sujeitos a desenvolver novas funções Neo-funcionalização Na prática Membros de uma mesma família de proteínas são homólogos e em geral tem mesma função Superfamílias e subfamílias
101 Família de proteínas Definição operacional Duas proteínas estão na mesma família se seus genes são homólogos ou (mais exigente) Duas proteínas estão na mesma família se seus genes são ortólogos Falar em proteínas homólogas é um certo abuso de linguagem: são os genes que são homólogos

102 Phylogenetic tree of the WHAMM proteins Kollmar et al. BMC Research Notes :88 doi: /
103 Colunas num AM devem ser homólogas O gene ancestral comum das sequências no AM também tinha aquela posição
104 Alinhar DNA ou aminoácidos? DNA: mais difícil garantir homologia nas colunas DNA é mais sensível, mas a 3a base de codons não é informativa Comparação com aminoácidos permite que proteínas mais distantes possam ser incluídas Há casos em que não dá para alinhar DNA (muita divergência) DNA é indicado quando as proteínas são todas idênticas ou quase idênticas Ex: cepas de uma bactéria 8/28/2018 J. C. Setubal 104
105 Algoritmo para alinhamento múltiplo de sequências Programação dinâmica Generalização de alinhamento 2-a-2 8/28/2018 J. C. Setubal 105

 O(n 3 ) Ω(2 k n k")
106 Generalização de PD para AM 2 sequências 3 sequências x y O(n 2 ) O(n 3 ) Ω(2 k n k )
107 Consequência Se PD para alinhamentos 2-a-2 já é caro para AM é ainda mais caro! Portanto todos os programas práticos para AM são heurísticas Não tem garantia de otimalidade (produzem aproximações)
108 Mesmo sendo heurísticas esses programas tem limitações As sequências de entrada: Não muito longas (menos do que ~10 kb) Não muitas (menos do que ~500) esses números variam dependendo do programa e do computador 8/28/2018 J. C. Setubal 108
109 Alinhamento progressivo Ideia: combinar alinhamentos de pares, iniciando com o par mais similar entre si Ir juntando os pares Dois estágios 1. constrói-se uma árvore-guia que determina a hierarquia de similaridade entre os pares 2. as sequências são adicionadas ao alinhamento num processo guiado pela árvore Seria melhor que AM e árvore fossem feitos simultaneamente Muito mais complicado de fazer com rigor
110 Programas para AM Muscle Edgar, R.C. (2004) Nucleic Acids Res. 32(5): MAFFT Katoh, Misawa, Kuma, Miyata 2002 (Nucleic Acids Res. 30: ) mafft.cbrc.jp/alignment/software/ ClustalW/X (antigos) Clustal Omega (novo) Sievers et al. Molecular Systems Biology (2011) 7: Outros: Probcons, Cobalt (NCBI), T-coffee 8/28/2018 J. C. Setubal 110
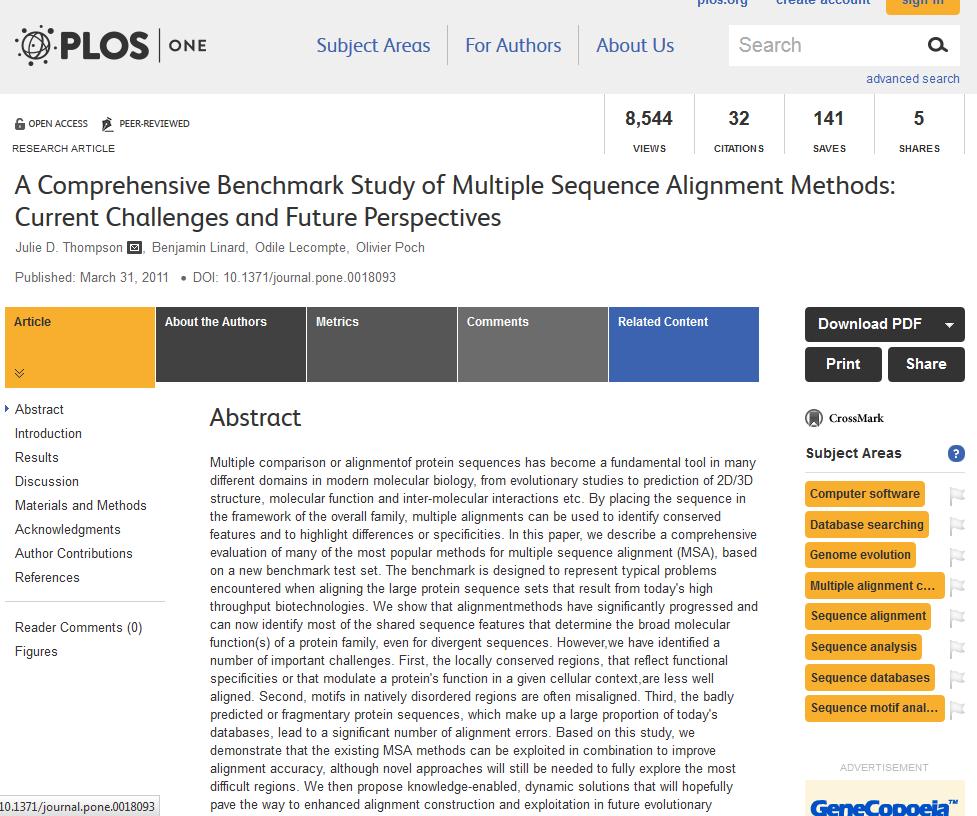
111
 Reference alignment of representative sequences of the p53/p63/p73 family, with the domain organization shown above (B) the alignment (AD: activation domain, Oligo: oligomerization, SAM: sterile")
112 Figure 1: An example benchmark alignment. (A) Reference alignment of representative sequences of the p53/p63/p73 family, with the domain organization shown above (B) the alignment (AD: activation domain, Oligo: oligomerization, SAM: sterile alpha motif). Colored blocks indicate conserved (C) regions. The grey regions correspond to sequence segments that could not be reliably aligned and white regions indicate (D) gaps in the alignment. (B) Different MSA programs produce different alignments, especially in the N-terminal region (boxe (E) d in red in A) containing rare motifs and a disordered proline-rich domain. Esquema comparativo de notas

113 Edição de alinhamentos Credit: R. Dixon 8/28/2018 J. C. Setubal 113
114 Edição de alinhamentos Algumas colunas podem não ser informativas No olho às vezes é possível ver alinhamentos locais melhores Edição manual Edição automática 8/28/2018 J. C. Setubal 114
115 Edição manual de AMs Jalview Waterhouse et al. Bioinformatics (9) Seaview Gouy M., Guindon S. & Gascuel O. (2010) Molecular Biology and Evolution 27(2):

116 JALVIEW 8/28/2018 J. C. Setubal 116
117
118 Edição automática de AMs GBLOCKS Castresana, J. (2000) Molecular Biology and Evolution 17, GUIDANCE Penn, O., Privman, E., Ashkenazy, H., Landan, G., Graur, D. and Pupko, T. (2010). GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Research, 2010 Jul 1; 38 (Web Server issue):w23-w28; doi: /nar/gkq443
119 Formatos de saída clustal, FASTA, MSF, NEXUS, PHYLIP
120 Alinhamento entre sequências longas Cromossomos inteiros O cromossomo típico de uma bactéria tem 4 Mbp Cromossomo de humanos: 300 Mbp Cromossomos e plasmídeos: replicons
121 28 August 2018 JC Setubal 121
122 BLAST não serve Computadores mesmo com dezenas de GB de RAM não dão conta de rodar BLAST para essas entradas Problema não é tempo; é memória RAM Outras abordagens são necessárias
123 O programa MUMmer Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res Jun 1;30(11): Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R August 2018 JC Setubal 123
124 Como MUMmer funciona It finds Maximal Unique Matches These are exact matches above a user-specified threshold that are unique Exact matches found are clustered and extended (using dynamic programming) Result is approximate matches Data structure for exact match finding: suffix tree Difficult to build but very fast Nucmer and promer Both very fast O(n + #MUMs), n = genome lengths 28 August 2018 JC Setubal 124
125 Árvore de sufixos para GTATCTAGG
126 Alinhamentos de replicons inteiros revelam rearranjos
127 Alinhamentos de pares de replicons completos Se as sequências fossem idênticas veríamos: B A 28 August 2018 JC Setubal 127
128 uma inversão A B C D A D C B 28 August 2018 JC Setubal 128
129 D B C A A B C D Such inversions seem to happen around 28 August 2018 the origin or terminus JC Setubalof replication 129
130 130
131 28 August 2018 JC Setubal 131
132
133 E. coli K12 Promer alignment Red: direct; green: reverse Xanthomonas axonopodis pv citri Both are γ proteobacteria! 28 August 2018 JC Setubal 133
134 Eisen JA, 28 August Heidelberg 2018 JF, White O, Salzberg SL. Evidence for JC symmetric Setubal chromosomal 134 inversions around the replication origin in bacteria. Genome Biol. 2000;1(6):RESEARCH0011
135 Alinhamento múltiplo de O programa MAUVE sequências longas Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res Jul;14(7): August 2018 JC Setubal 135
136 How MAUVE works Seed-and-extend hashing Seeds/anchors: Maximal Multiple Unique Matches of minimum length k Result: Local collinear blocks (LCBs) O(G 2 n + Gn log Gn), G = # genomes, n = average genome length 28 August 2018 JC Setubal 136
137 Alignment algorithm 1. Find Multi-MUMs 2. Use the multi-mums to calculate a phylogenetic guide tree 3. Find LCBs (subset of multi-mums; filter out spurious matches; requires minimum weight) 4. Recursive anchoring to identify additional anchors (extension of LCBs) 5. Progressive alignment (CLUSTALW) using guide tree 28 August 2018 JC Setubal 137

138 Brucella: Main chromosome alignment 28 August 2018 JC Setubal 138

139 Brucella: Chromosome 2 alignment 28 August 2018 JC Setubal 139

140 Coxiella: Chromosome alignment Dugway RSA 493 RSA August 2018 JC Setubal 140

141 Rickettsia 28 August 2018 JC Setubal 141
142 Sumário de comparação de sequências Sequências curtas Sequências longas 2-a-2 Prog. Dinâmica Mummer 2-a-2 muitas vezes BLAST, usearch, diamond Mummer múltiplo Muscle, MAFFT Mauve, MUGSY
143 Distância genômica Ao comparar genomas, muitas vezes é útil poder expressar essa comparação por meio de um único número Quando se comparam pares de replicons Que podem ser replicons concatenados Distância pode ser entendida como o inverso da similaridade
144 Distância e similaridade São conceitos muito parecidos Em particular distância de edição Como transformar sequência s em sequência t Operações Substituição do caracter a por b (custo = 1) Inserção ou Remoção de um caracter (custo = 2) O algoritmo de PD já visto resolve esse problema 8/28/2018 J. C. Setubal 144
145 Uma fórmula de distância MUMi = MUM index Baseado em MUMmer Deloger et al genômica MUMi = 1 L mum /L av L mum = soma dos comprimentos de todos os MUMs que não tem sobreposição L av = comprimento médio dos 2 genomas sendo comparados Para obter MUMi, basta rodar MUMmer com um script perl desses autores 8/28/2018 J. C. Setubal 145
146 How MUMi works Identical sequences: zero Totally different sequences: 1 The boundary between genus and species is around 0.8

147 Distribution of all minimal MUMi values per genus. Marc Deloger et al. J. Bacteriol. 2009;191:91-99
148 Conclusão Não dá para comparar distâncias MUMi entre diferentes gêneros 8/28/2018 J. C. Setubal 148
149 Uma matriz de distâncias genômicas em Brucella 8/28/2018 J. C. Setubal 149

150 Valores MUMi em Xanthomonas
 For comparison: distance between Stenotrophomonas")
151 Largest distance: (X. perforans and X. fuscans aurantifolii C 1559) For comparison: distance between Stenotrophomonas maltophilia and Xac 306: 0.912
152 ANI Average Nucleotide Identity [Goris et al. 2007] Baseado em BLAST one-way ANI (best hits) two-way ANI (reciprocal best hits) Typically, 2 species will have ANI 95% < 75% : not to be trusted
153 GDDC Genome-to-genome distance calculation rigorous in silico replacement for DNA-DNA hybridization wet-lab experiments Meier-Kolthoff, J.P., et al., Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics, : p. 60
154 Comparação de conjuntos de genes Given a set of genomes, represented by their proteomes or sets of protein sequences Given homologous relationships (as given for example by orthomcl) Which genes are shared by genomes X and Y? Which genes are unique to genome Z? Venn or extended Venn diagrams 28 August 2018 JC Setubal 154
155 3-way genome comparison A B C 28 August 2018 JC Setubal 155
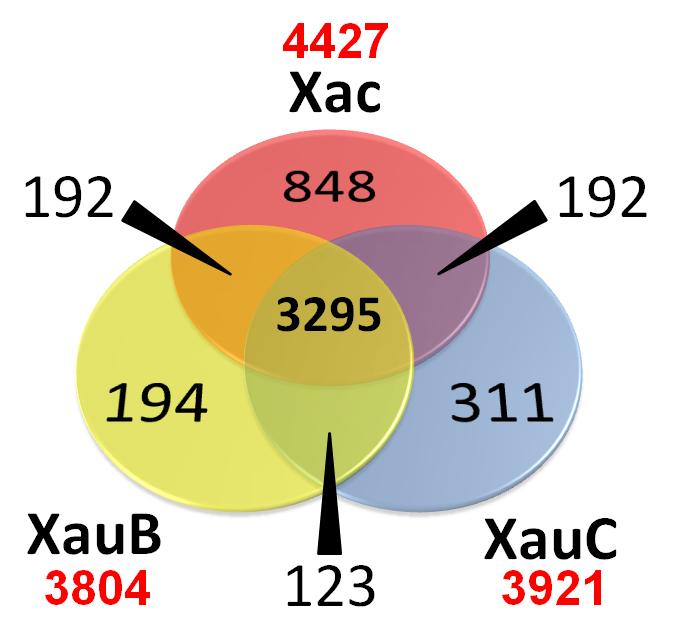
156


157 Diagrama de Venn para n = 6 Número de comparações é quadrático em n Número de regiões num diagrama de Venn = 2 n

158 Wulff et al. MPMI Vol. 27, No. 2, 2014, pp
159 Cômputo de famílias de proteínas 1. Verificar as similaridades entre as sequências a) Usando (por exemplo) BLAST + critérios 2. Representar as similaridades num grafo 3. Aplicar um algoritmo de clusterização sobre o grafo
160 Clusterização é necessária porque o grafo pode ser complexo a b c
161 Resultado da clusterização Matriz com genes nas colunas (g j ), genomas nas linhas (O i ) Cada coluna representa uma família de genes homólogos g1 g2 g3 g4 O1 O2 O3 O4 O5 O6
162 orthomcl pipeline Li Li et al. Genome Res. 2003; 13:
163
164
165
166 Pan genoma; genoma core e genoma acessório A B core C pan: A U B U C acessório: pan - core 28 August 2018 JC Setubal 166
167 Curva de pan-genoma (n = 4)
168 Curva de core genoma (n = 4) O número de genes para x=1 não é o mesmo do gráfico anterior pois os singletons são descartados
169 Genomas fechados e abertos Fechado O número de genes/famílias do pan-genoma atinge um platô Aberto O número de genes/famílias não atinge um platô; cresce sempre que se acrescentam novos genomas
170
171 O genoma de E. coli é aberto Fonte: pangp.big.ac.cn
172 Bacillus anthracis Genoma fechado Cerca de genes
173 Ferramentas para análise pan/core Get_Homologues Contreras-Moreira, B. and P. Vinuesa, GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol, (24): p Roary Page, A.J., et al., Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics, (22): p
174 Conjuntos + contexto Como genes compartilhados aparecem em seus respectivos genomas? Filogenômica Busca de sintenia = preservação de ordem Basta fazer um alinhamento Os caracteres a serem alinhados são os genes

175 Alinhamento de ortólogos obtido no IMG/JGI

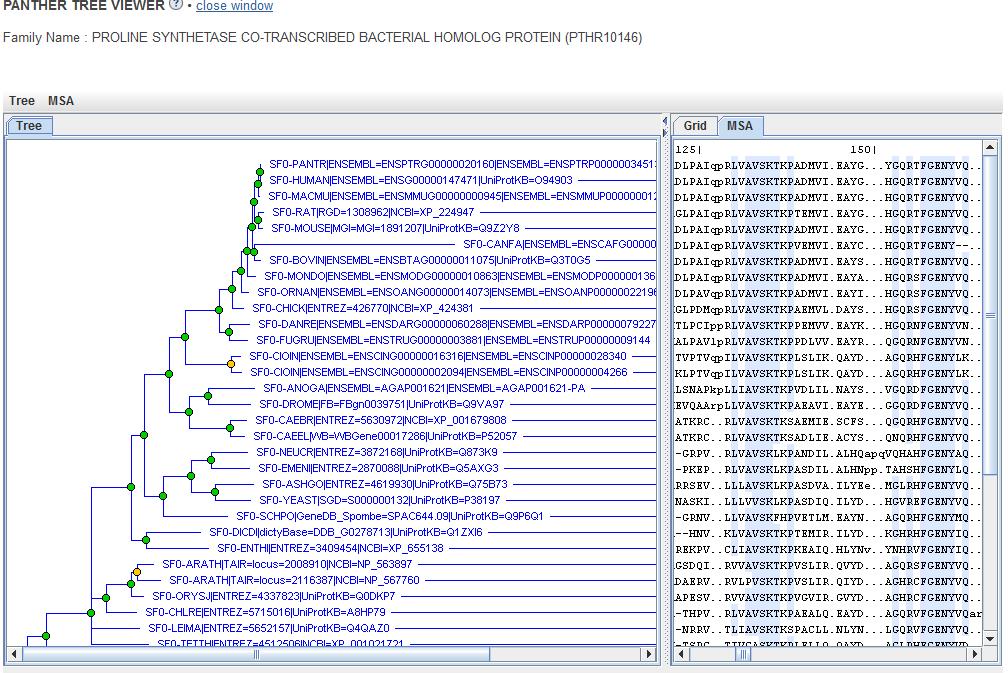
176 Roda da ortologia
177
178 Sumário - 1 Comparação de sequências curtas 2-a-2 Alinhamento Sistemas de pontuação Matrizes de substituição Programação dinâmica Significância estatística de alinhamentos BLAST
179 Sumário - 2 Comparação de várias sequências ao mesmo tempo Alinhamento múltiplo Programas Comparação de sequências longas 2-a-2 Alinhamento múltiplo
180 Sumário - 3 Distâncias genômicas Comparação entre conjuntos de genes Pan/Core Sintenia
 KEGG")
181 Protein family resources Clusters of orthologous groups (COG, KOG, eggnog) KEGG orthologs

182

183 Query by accession
184
185
186
187
188

189 Phylofacts query by sequence search

190 Resource federation: InterPro
191 Not as easy as it may sound Specific protein families may not be consistent across resources Most families (MSAs, trees, HMMs) in these resources are not manually curated Domains in Pfam-A are curated TIGRfams are curated HAMAP families are curated

192
193
194 proteômica
195 Fig. 4. Net gene loss or gain throughout the evolution of the {alpha}-proteobacterial species Boussau, Bastien et al. (2004) Proc. Natl. Acad. Sci. USA 101, August 2018 JC Setubal 195 Copyright 2004 by the National Academy of Sciences
Comparação de sequências
 Comparação de sequências João Carlos Setubal IQ-USP 2013 9/26/2013 J. C. Setubal 1 Referência J.C. Setubal Chapter A05 Similarity Search http://www.ncbi.nlm.nih.gov/books/nbk6831 Motivação: Por que comparar
Comparação de sequências João Carlos Setubal IQ-USP 2013 9/26/2013 J. C. Setubal 1 Referência J.C. Setubal Chapter A05 Similarity Search http://www.ncbi.nlm.nih.gov/books/nbk6831 Motivação: Por que comparar
Comparação de sequências
 Comparação de sequências João Carlos Setubal IQ-USP 2014 9/18/2014 J. C. Setubal 1 Esta aula está em www.iq.usp.br/setubal/bmc/2013/bmc.html Alinhamentos e comparações Referência J.C. Setubal Chapter A05
Comparação de sequências João Carlos Setubal IQ-USP 2014 9/18/2014 J. C. Setubal 1 Esta aula está em www.iq.usp.br/setubal/bmc/2013/bmc.html Alinhamentos e comparações Referência J.C. Setubal Chapter A05
Comparação de sequências
 Comparação de sequências João Carlos Setubal IQ-USP 2016 9/22/2016 J. C. Setubal 1 Motivação Ciccarelli et al, Science, 2006 Como foi possível criar tal árvore? Determinar genes que são compartilhados
Comparação de sequências João Carlos Setubal IQ-USP 2016 9/22/2016 J. C. Setubal 1 Motivação Ciccarelli et al, Science, 2006 Como foi possível criar tal árvore? Determinar genes que são compartilhados
Comparação de sequências
 Comparação de sequências João Carlos Setubal IQ-USP 2017 9/25/2017 J. C. Setubal 1 Motivação Ciccarelli et al, Science, 2006 Como foi possível criar tal árvore? Determinar genes que são compartilhados
Comparação de sequências João Carlos Setubal IQ-USP 2017 9/25/2017 J. C. Setubal 1 Motivação Ciccarelli et al, Science, 2006 Como foi possível criar tal árvore? Determinar genes que são compartilhados
Principais algoritmos de alinhamento de sequências genéticas. Alexandre dos Santos Cristino
 Principais algoritmos de alinhamento de sequências genéticas Alexandre dos Santos Cristino http://www.ime.usp.br/~alexsc e-mail: alexsc@ime.usp.br Definição de alinhamento de sequências Comparação de duas
Principais algoritmos de alinhamento de sequências genéticas Alexandre dos Santos Cristino http://www.ime.usp.br/~alexsc e-mail: alexsc@ime.usp.br Definição de alinhamento de sequências Comparação de duas
Métodos de alinhamento de sequências biológicas. Marcelo Falsarella Carazzolle
 Métodos de alinhamento de sequências biológicas Marcelo Falsarella Carazzolle Resumo - Introdução - Alinhamentos ótimos - Global - Local (Smith-Waterman) - Semi global - Matrizes de alinhamento (BLOSUM)
Métodos de alinhamento de sequências biológicas Marcelo Falsarella Carazzolle Resumo - Introdução - Alinhamentos ótimos - Global - Local (Smith-Waterman) - Semi global - Matrizes de alinhamento (BLOSUM)
Alinhamentos e Busca de Similaridade. Ariane Machado Lima
 Alinhamentos e Busca de Similaridade Ariane Machado Lima Busca de identidade Identificar o que é determinada seqüência Ex.acabou de seqüenciar, seria contaminante? Outras fases de um projeto de seqüenciamento
Alinhamentos e Busca de Similaridade Ariane Machado Lima Busca de identidade Identificar o que é determinada seqüência Ex.acabou de seqüenciar, seria contaminante? Outras fases de um projeto de seqüenciamento
Alinhamentos de sequências e Busca de Similaridade
 Alinhamentos de sequências e Busca de Similaridade Ariane Machado Lima ariane.machado@usp.br Escola de Artes, Ciências e Humanidades - USP Contexto http://www.ekac.org/gene.html http://www.fuzzco.com/news/wp-content/uploads/27//genome.jpg
Alinhamentos de sequências e Busca de Similaridade Ariane Machado Lima ariane.machado@usp.br Escola de Artes, Ciências e Humanidades - USP Contexto http://www.ekac.org/gene.html http://www.fuzzco.com/news/wp-content/uploads/27//genome.jpg
Alinhamento de sequências
 Pontifícia Universidade Católica de Goiás Departamento de Biologia Alinhamento de sequências Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com Definição O alinhamento de sequências consiste no
Pontifícia Universidade Católica de Goiás Departamento de Biologia Alinhamento de sequências Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com Definição O alinhamento de sequências consiste no
Alinhamento de seqüências
 Alinhamento de seqüências Qual a importância do alinhamento de seqüências Permite estabelecer identidades entre sequências Permite a dedução de função de proteínas baseado em similaridade Permite a definição
Alinhamento de seqüências Qual a importância do alinhamento de seqüências Permite estabelecer identidades entre sequências Permite a dedução de função de proteínas baseado em similaridade Permite a definição
Alinhamento local- Utilização do BLAST
 Alinhamento local- Utilização do BLAST BLAST Tipos de BLAST (blastn) Compara nucleotídeos (blastp) Compara proteínas Utiliza nucleotídeo como query, este é traduzido nos seus 6 quadros de leitura e é comparado
Alinhamento local- Utilização do BLAST BLAST Tipos de BLAST (blastn) Compara nucleotídeos (blastp) Compara proteínas Utiliza nucleotídeo como query, este é traduzido nos seus 6 quadros de leitura e é comparado
Tópicos Especiais em Inteligência Artificial COS746. Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro
 Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven/C. David Page para BMI/CS 576,
Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven/C. David Page para BMI/CS 576,
Tópicos Especiais em Inteligência Artificial COS746. Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro
 Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven para BMI/CS 576, UW-Madison
Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven para BMI/CS 576, UW-Madison
Nada em Biologia faz sentido senão à luz da evolução.
 Marcos T. Geraldo ADAPTABILIDADE Nada em Biologia faz sentido senão à luz da evolução. Theodosius Dobzhansky (1973) 1 Processo de evolução em moléculas de DNA, RNA e proteínas Reconstrução das relações
Marcos T. Geraldo ADAPTABILIDADE Nada em Biologia faz sentido senão à luz da evolução. Theodosius Dobzhansky (1973) 1 Processo de evolução em moléculas de DNA, RNA e proteínas Reconstrução das relações
alinhamento global-alinhamento múltiplo de seqüências
 alinhamento global-alinhamento múltiplo de seqüências Alinhamento múltiplos de seqüências Qual a importância de se realizar alinhamentos múltiplos em oposição a alinhamentos em pares? Alinhamento múltiplos
alinhamento global-alinhamento múltiplo de seqüências Alinhamento múltiplos de seqüências Qual a importância de se realizar alinhamentos múltiplos em oposição a alinhamentos em pares? Alinhamento múltiplos
Análise de significância de. alinhamentos
 Análise de significância de alinhamentos Análise de significância de um alinhamento Tão importante como escolher o método de scoring ou encontrar o alinhamento que maximiza o score é saber avaliar a significância
Análise de significância de alinhamentos Análise de significância de um alinhamento Tão importante como escolher o método de scoring ou encontrar o alinhamento que maximiza o score é saber avaliar a significância
Alinhamentos de Múltiplas Seqüências. Rogério T. Brito Orientador: José A. R. Soares
 1 Alinhamentos de Múltiplas Seqüências Rogério T. Brito Orientador: José A. R. Soares 2 Motivação Problema em Biologia: saber qual é o grau de parentesco entre um conjunto de espécies (construção de árvores
1 Alinhamentos de Múltiplas Seqüências Rogério T. Brito Orientador: José A. R. Soares 2 Motivação Problema em Biologia: saber qual é o grau de parentesco entre um conjunto de espécies (construção de árvores
Biologia Molecular Computacional Homologia
 Biologia Molecular Computacional Homologia Luiz Thibério Rangel O que é homologia? Conceito básico para estudos de genômica comparativa; Passo inicial para estudos de filogenia(omica); Importante para
Biologia Molecular Computacional Homologia Luiz Thibério Rangel O que é homologia? Conceito básico para estudos de genômica comparativa; Passo inicial para estudos de filogenia(omica); Importante para
TITULO: Implementação do alinhamento de proteínas em GPU utilizando OpenCL PROPOSTA DE TRABALHO DE GRADUAÇÃO
 1 U NIVERSIDADE FEDERAL DE PERNAMBUCO GRADUAÇÃO EM ENGENHARIA DA COMPUTAÇÃO CENTRO DE INFORMÁTICA 2 0 1 6. 1 TITULO: Implementação do alinhamento de proteínas em GPU utilizando OpenCL PROPOSTA DE TRABALHO
1 U NIVERSIDADE FEDERAL DE PERNAMBUCO GRADUAÇÃO EM ENGENHARIA DA COMPUTAÇÃO CENTRO DE INFORMÁTICA 2 0 1 6. 1 TITULO: Implementação do alinhamento de proteínas em GPU utilizando OpenCL PROPOSTA DE TRABALHO
Alinhamento de Sequências e Genômica Comparativa
 Encontro França-Brasil de Bioinformática Universidade Estadual de Santa Cruz (UESC) Ilhéus-BA - Brasil Alinhamento de Sequências e Genômica Comparativa Maria Emília M. T. Walter Departamento de Ciência
Encontro França-Brasil de Bioinformática Universidade Estadual de Santa Cruz (UESC) Ilhéus-BA - Brasil Alinhamento de Sequências e Genômica Comparativa Maria Emília M. T. Walter Departamento de Ciência
Identificação de genes por similaridade de seqüência
 Identificação de genes por similaridade de seqüência Evolução do genoma Os genes evoluem a partir de genes ancestrais comuns acumulando mutações Homologia Genes ancestrais estão presentes nas espécies
Identificação de genes por similaridade de seqüência Evolução do genoma Os genes evoluem a partir de genes ancestrais comuns acumulando mutações Homologia Genes ancestrais estão presentes nas espécies
Programas de Alinhamento. Sumário
 Programas de Alinhamento Departamento de Genética FMRP- USP Alynne Oya Chiromatzo alynne@lgmb.fmrp.usp.br Sumário Introdução para buscas em base de dados Fasta Blast Programa para alinhamento Clustal 1
Programas de Alinhamento Departamento de Genética FMRP- USP Alynne Oya Chiromatzo alynne@lgmb.fmrp.usp.br Sumário Introdução para buscas em base de dados Fasta Blast Programa para alinhamento Clustal 1
Bioinformática para o Citrus EST Project (CitEST)
") Bioinformática para o Citrus EST Project (CitEST) Marcelo da Silva Reis 1 1 Instituto de Matemática e Estatística, Universidade de São Paulo 20 de maio de 2009 Organização da Apresentação Esta apresentação
Bioinformática para o Citrus EST Project (CitEST) Marcelo da Silva Reis 1 1 Instituto de Matemática e Estatística, Universidade de São Paulo 20 de maio de 2009 Organização da Apresentação Esta apresentação
Bioinformática. João Varela Aulas T7-T8 CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, E ENGENHARIA BIOLÓGICA
 Bioinformática CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, E ENGENHARIA BIOLÓGICA João Varela jvarela@ualg.pt Aulas T7-T8 Esquema de anotação Annothaton 1. Determinar a localização das ORFs presentes
Bioinformática CURSOS EM BIOLOGIA, BIOQUÍMICA, BIOTECNOLOGIA, E ENGENHARIA BIOLÓGICA João Varela jvarela@ualg.pt Aulas T7-T8 Esquema de anotação Annothaton 1. Determinar a localização das ORFs presentes
PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE
 CONCEITOS EM EPIDEMIOLOGIA E FILOGENIA MOLECULARES PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE DE
CONCEITOS EM EPIDEMIOLOGIA E FILOGENIA MOLECULARES PAULO EDUARDO BRANDÃO, PhD DEPARTAMENTO DE MEDICINA VETERINÁRIA PREVENTIVA E SAÚDE ANIMAL FACULDADE DE MEDICINA VETERINÁRIA E ZOOTECNIA UNIVERSIDADE DE
A matemática e o genoma. Resumo
 I Coloquio Regional da Região Centro-Oeste, 3 a 6 de novembro de 2009 Universidade Federal de Mato Grosso do Sul Mini-curso A matemática e o genoma Nalvo F. Almeida Jr. Resumo Os avanços da biotecnologia
I Coloquio Regional da Região Centro-Oeste, 3 a 6 de novembro de 2009 Universidade Federal de Mato Grosso do Sul Mini-curso A matemática e o genoma Nalvo F. Almeida Jr. Resumo Os avanços da biotecnologia
Elisa Boari de Lima Orientador: Thiago de Souza Rodrigues
 Uma Metodologia para Identificação de Módulos Formadores de Sequências de Proteínas Mosaicas do Trypanosoma cruzi a partir do Transcriptoma do Parasito Utilizando a Ferramenta BLAST Elisa Boari de Lima
Uma Metodologia para Identificação de Módulos Formadores de Sequências de Proteínas Mosaicas do Trypanosoma cruzi a partir do Transcriptoma do Parasito Utilizando a Ferramenta BLAST Elisa Boari de Lima
Bases de Dados. Freqüentemente usadas em. Bioinformática
 Bases de Dados Freqüentemente usadas em Bioinformática Ana Carolina Q. Simões anakqui@yahoo.com Organização da aula NCBI Translate tool Genome Browser EBI SwissProt KEGG Gene Ontology SMD Revistas relevantes
Bases de Dados Freqüentemente usadas em Bioinformática Ana Carolina Q. Simões anakqui@yahoo.com Organização da aula NCBI Translate tool Genome Browser EBI SwissProt KEGG Gene Ontology SMD Revistas relevantes
Marcelo Reis. Centro APTA Citros Sylvio Moreira. 18 de julho de 2007
 I n t r o d u ç ã o à B i o i n f o r m á t i c a Marcelo Reis Centro APTA Citros Sylvio Moreira 18 de julho de 2007 Duração estimada: ~ 2,5h (manhã) ~ 2,5h (tarde) A g e n d a Manhã: Que trem é esse,
I n t r o d u ç ã o à B i o i n f o r m á t i c a Marcelo Reis Centro APTA Citros Sylvio Moreira 18 de julho de 2007 Duração estimada: ~ 2,5h (manhã) ~ 2,5h (tarde) A g e n d a Manhã: Que trem é esse,
Teoria dos Grafos Aula 17
 Teoria dos Grafos Aula 17 Aula passada Problema da soma do subconjunto (subset sum) Programação dinâmica Problema da mochila Aula de hoje Alinhamento de sequências Programação dinâmica Caminho mais curto
Teoria dos Grafos Aula 17 Aula passada Problema da soma do subconjunto (subset sum) Programação dinâmica Problema da mochila Aula de hoje Alinhamento de sequências Programação dinâmica Caminho mais curto
Busca de motivos em sequências. João Carlos Setubal IQ-USP 2014
 Busca de motivos em sequências João Carlos Setubal IQ-USP 2014 Motivos do tipo I AACT(G A)N 12 AGTT Q-[LIV]-H-H-[SA]-x(2)-D-G-[FY]-H Chloramphenicol acetyltransferase active site (do PROSITE) Posições
Busca de motivos em sequências João Carlos Setubal IQ-USP 2014 Motivos do tipo I AACT(G A)N 12 AGTT Q-[LIV]-H-H-[SA]-x(2)-D-G-[FY]-H Chloramphenicol acetyltransferase active site (do PROSITE) Posições
Aminoácidos. Prof. Dr. Walter F. de Azevedo Jr. Laboratório de Sistemas BioMoleculares. Departamento de Física. UNESP São José do Rio Preto. SP.
 Aminoácidos Prof. Dr. Walter F. de Azevedo Jr. Laboratório de Sistemas BioMoleculares. Departamento de Física. UNESP São José do Rio Preto. SP. Resumo Introdução Quiralidade Ligação peptídica Cadeia peptídica
Aminoácidos Prof. Dr. Walter F. de Azevedo Jr. Laboratório de Sistemas BioMoleculares. Departamento de Física. UNESP São José do Rio Preto. SP. Resumo Introdução Quiralidade Ligação peptídica Cadeia peptídica
2 Contexto e Motivações
 2 Contexto e Motivações Existem duas principais famílias de algoritmos que realizam comparações de biosseqüências, a FASTA (Pearson, 1991) e a BLAST (Altschul et al., 1990). Estas ferramentas realizam
2 Contexto e Motivações Existem duas principais famílias de algoritmos que realizam comparações de biosseqüências, a FASTA (Pearson, 1991) e a BLAST (Altschul et al., 1990). Estas ferramentas realizam
Busca de motivos em sequências. João Carlos Setubal 2015
 Busca de motivos em sequências João Carlos Setubal 2015 Cadeias exatas Podem ser encontradas com o mecanismo de busca de qualquer editor de textos Que algoritmo é executado? O mais simples (e que é muito
Busca de motivos em sequências João Carlos Setubal 2015 Cadeias exatas Podem ser encontradas com o mecanismo de busca de qualquer editor de textos Que algoritmo é executado? O mais simples (e que é muito
Alinhamento de Seqüências
 18 CAPÍTULO 3 Alinhamento de Seqüências 3.1. Introdução O alinhamento de seqüências consiste no processo de comparar duas seqüências (de nucleotídeos ou proteínas) de forma a se observar seu nível de identidade.
18 CAPÍTULO 3 Alinhamento de Seqüências 3.1. Introdução O alinhamento de seqüências consiste no processo de comparar duas seqüências (de nucleotídeos ou proteínas) de forma a se observar seu nível de identidade.
Protein Homology detection by HMM-comparation.
 UNIVERSIDADE FEDERAL DE PERNAMBUCO Cin Centro de Informática Pós-Graduação em Ciência da Computação Protein Homology detection by HMM-comparation. Johannes Soding Vol. 21 no. 7 2005, BIOINFORMATICS Recife,
UNIVERSIDADE FEDERAL DE PERNAMBUCO Cin Centro de Informática Pós-Graduação em Ciência da Computação Protein Homology detection by HMM-comparation. Johannes Soding Vol. 21 no. 7 2005, BIOINFORMATICS Recife,
Bioinformática. Licenciaturas em Biologia, Bioquímica, Biotecnologia, Engenharia Biológica. João Varela
 Bioinformática Licenciaturas em Biologia, Bioquímica, Biotecnologia, Engenharia Biológica João Varela jvarela@ualg.pt Docentes João Varela (bioinformática: conceitos, bases de dados, aplicações, pesquisa
Bioinformática Licenciaturas em Biologia, Bioquímica, Biotecnologia, Engenharia Biológica João Varela jvarela@ualg.pt Docentes João Varela (bioinformática: conceitos, bases de dados, aplicações, pesquisa
Número: Nome:
 Número: Nome: 1 -------------------------------------------------------------------------------------------------------------- INSTITUTO SUPERIOR TÉCNICO Sistemas de Apoio à Decisão Exame 1 20 junho 2006
Número: Nome: 1 -------------------------------------------------------------------------------------------------------------- INSTITUTO SUPERIOR TÉCNICO Sistemas de Apoio à Decisão Exame 1 20 junho 2006
Comparação e alinhamento de. sequências
 Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Ancoragem de genomas incompletos em genomas completos
 Ancoragem de genomas incompletos em genomas completos André Chastel Lima Dissertação de Mestrado Orientação: Prof. Dr. Nalvo Franco de Almeida Junior Área de Concentração: Biologia Computacional Dissertação
Ancoragem de genomas incompletos em genomas completos André Chastel Lima Dissertação de Mestrado Orientação: Prof. Dr. Nalvo Franco de Almeida Junior Área de Concentração: Biologia Computacional Dissertação
Árvore Binária de Busca Ótima
 MAC 5710 - Estruturas de Dados - 2008 Referência bibliográfica Os slides sobre este assunto são parcialmente baseados nas seções sobre árvore binária de busca ótima do capítulo 4 do livro N. Wirth. Algorithms
MAC 5710 - Estruturas de Dados - 2008 Referência bibliográfica Os slides sobre este assunto são parcialmente baseados nas seções sobre árvore binária de busca ótima do capítulo 4 do livro N. Wirth. Algorithms
Identificação de Padrões em Proteínas Utilizando a Ferramenta de Bioinformática CD- Search
 4ª Jornada Científica e Tecnológica e 1º Simpósio de Pós-Graduação do IFSULDEMINAS 16, 17 e 18 de outubro de 2012, Muzambinho MG Identificação de Padrões em Proteínas Utilizando a Ferramenta de Bioinformática
4ª Jornada Científica e Tecnológica e 1º Simpósio de Pós-Graduação do IFSULDEMINAS 16, 17 e 18 de outubro de 2012, Muzambinho MG Identificação de Padrões em Proteínas Utilizando a Ferramenta de Bioinformática
Transcritômica. João Carlos Setubal IQ/USP outubro de 2013
 Transcritômica João Carlos Setubal IQ/USP outubro de 2013 Objetivo Obter, analisar, e interpretar dados de expressão gênica mrnas (que vão virar proteína) RNAs (que não vão virar proteína; ncrnas) O gene
Transcritômica João Carlos Setubal IQ/USP outubro de 2013 Objetivo Obter, analisar, e interpretar dados de expressão gênica mrnas (que vão virar proteína) RNAs (que não vão virar proteína; ncrnas) O gene
ANÁLISE DE COMPLEXIDADE DOS ALGORITMOS
 1/18 ANÁLISE DE COMPLEXIDADE DOS ALGORITMOS Algoritmos 2/18 Algoritmos Algoritmo - sequência de instruções necessárias para a resolução de um problema bem formulado (passíveis de implementação em computador)
1/18 ANÁLISE DE COMPLEXIDADE DOS ALGORITMOS Algoritmos 2/18 Algoritmos Algoritmo - sequência de instruções necessárias para a resolução de um problema bem formulado (passíveis de implementação em computador)
ALINHAMENTO DE SEQUÊNCIAS
 Disciplina de BIOLOGIA COMPUTACIONAL Mestrado em ENGENHARIA BIOMÉDICA 4º Ano, 1º Semestre 2007/08 ALINHAMENTO DE SEQUÊNCIAS Relatório 2 Ana Calhau Ângela Pisco Nuno Santos 54605 55748 55746 Palavras-Chave:
Disciplina de BIOLOGIA COMPUTACIONAL Mestrado em ENGENHARIA BIOMÉDICA 4º Ano, 1º Semestre 2007/08 ALINHAMENTO DE SEQUÊNCIAS Relatório 2 Ana Calhau Ângela Pisco Nuno Santos 54605 55748 55746 Palavras-Chave:
Comparação entre sequências biológicas
 Comparação entre sequências biológicas Extraíndo e analisando os sinais contidos em biopolímeros ( Alinhamentos ) Prof. Dr. Alessandro Varani UNESP - FCAV Objetivos Abordagens práticas para comparação
Comparação entre sequências biológicas Extraíndo e analisando os sinais contidos em biopolímeros ( Alinhamentos ) Prof. Dr. Alessandro Varani UNESP - FCAV Objetivos Abordagens práticas para comparação
Genômica. Desenvolvimento e Aplicações. Prof. Manoel Victor
 Genômica Desenvolvimento e Aplicações Definições Genoma: informações do complemento genético de um indivíduo ou de sua espécie freqüentemente entendido como a seqüência de nucleotídeos do genoma Genômica:
Genômica Desenvolvimento e Aplicações Definições Genoma: informações do complemento genético de um indivíduo ou de sua espécie freqüentemente entendido como a seqüência de nucleotídeos do genoma Genômica:
Busca de motivos em sequências. João Carlos Setubal IQ-USP 2016
 Busca de motivos em sequências João Carlos Setubal IQ-USP 2016 Cadeias exatas Podem ser encontradas com o mecanismo de busca de qualquer editor de textos Que algoritmo é executado? O mais simples (e que
Busca de motivos em sequências João Carlos Setubal IQ-USP 2016 Cadeias exatas Podem ser encontradas com o mecanismo de busca de qualquer editor de textos Que algoritmo é executado? O mais simples (e que
Anotação de Genomas. Fabiana G. S. Pinto
 Anotação de Genomas Fabiana G. S. Pinto Obtenção de Seqüências geradas pelo MegaBace 1000 Dados brutos (medidas analógicas) de saída do seqüênciamento Base calling BIOINFORMÁTICA * PHRED: - Transforma
Anotação de Genomas Fabiana G. S. Pinto Obtenção de Seqüências geradas pelo MegaBace 1000 Dados brutos (medidas analógicas) de saída do seqüênciamento Base calling BIOINFORMÁTICA * PHRED: - Transforma
Algoritmos 3/17/ Algoritmos como área de estudo e investigação
 Algoritmos e Complexidade Ana Teresa Freitas INESC-ID/IST ID/IST 3/17/2005 1 O que é um algoritmo? Algoritmos: Sequência de instruções necessárias para a resolução de um problema bem formulado [passíveis
Algoritmos e Complexidade Ana Teresa Freitas INESC-ID/IST ID/IST 3/17/2005 1 O que é um algoritmo? Algoritmos: Sequência de instruções necessárias para a resolução de um problema bem formulado [passíveis
BANCO DE DADOS BIOLÓGICOS Aula 11
 BANCO DE DADOS BIOLÓGICOS Aula 11 Estudo dirigido 1. O que fazer com uma sequência de DNA? 2. Bancos de dados públicos e internacionais: GenBank, ENA, DDBJ; 3. NCBI; EMBL; DDBJ; 4. Sequências completas
BANCO DE DADOS BIOLÓGICOS Aula 11 Estudo dirigido 1. O que fazer com uma sequência de DNA? 2. Bancos de dados públicos e internacionais: GenBank, ENA, DDBJ; 3. NCBI; EMBL; DDBJ; 4. Sequências completas
Comparação e alinhamento de sequências
 Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Comparação e alinhamento de sequências Comparar sequências A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo
Dados Moleculares x Morfológicos
 Evolução Molecular Dados Moleculares x Morfológicos Hereditários Descrição não ambígua Mais fácil estabelecer homologia Permite comparações de espécies distantes Abundantes Fatores ambientais Diferenças
Evolução Molecular Dados Moleculares x Morfológicos Hereditários Descrição não ambígua Mais fácil estabelecer homologia Permite comparações de espécies distantes Abundantes Fatores ambientais Diferenças
Metagenômica. João Carlos Setubal IQ/USP
 Metagenômica João Carlos Setubal IQ/USP Estudo de genomas isolados Isolar organismo Cultivar Extrair DNA Fragmentar DNA Sequenciar os fragmentos DNA Montar os fragmentos bioinformática Anotar a sequência
Metagenômica João Carlos Setubal IQ/USP Estudo de genomas isolados Isolar organismo Cultivar Extrair DNA Fragmentar DNA Sequenciar os fragmentos DNA Montar os fragmentos bioinformática Anotar a sequência
Capítulo 8. Versão 0.4. Filogenômica
 Capítulo 8 Versão 0.4 Filogenômica O termo "Filogenômica" é definido de várias maneiras, mas, em geral, a definição é relacionada com a intersecção dos campos da Genômica e da evolução biológica. Aqui,
Capítulo 8 Versão 0.4 Filogenômica O termo "Filogenômica" é definido de várias maneiras, mas, em geral, a definição é relacionada com a intersecção dos campos da Genômica e da evolução biológica. Aqui,
Explorando bancos de dados genômicos e introdução à bioinformática. Guilherme Targino Valente Marcos Tadeu Geraldo. Bioinformática
 Explorando bancos de dados genômicos e introdução à bioinformática Guilherme Targino Valente Marcos Tadeu Geraldo 22/07/2011 Bioinformática É a aplicação de estatística e ciência da computação no campo
Explorando bancos de dados genômicos e introdução à bioinformática Guilherme Targino Valente Marcos Tadeu Geraldo 22/07/2011 Bioinformática É a aplicação de estatística e ciência da computação no campo
Explorando genomas: predição de genes e elementos transponíveis Proporção de diferentes sequências no genoma
 Explorando genomas: predição de genes e elementos transponíveis Proporção de diferentes sequências no genoma 1 Especies Genoma Genes 11 O número de genes varia entre as espécies 2 Anotação do Genoma 1
Explorando genomas: predição de genes e elementos transponíveis Proporção de diferentes sequências no genoma 1 Especies Genoma Genes 11 O número de genes varia entre as espécies 2 Anotação do Genoma 1
UFPel CDTec Biotecnologia. Anotação de genomas. MSc. Frederico schmitt Kremer
 UFPel CDTec Biotecnologia Anotação de genomas MSc. Frederico schmitt Kremer A anotação de um genoma consiste na identificação de suas regiões funcionais ou de relevância biológico, o que pode incluir:
UFPel CDTec Biotecnologia Anotação de genomas MSc. Frederico schmitt Kremer A anotação de um genoma consiste na identificação de suas regiões funcionais ou de relevância biológico, o que pode incluir:
Turma de terça-feira 14 hs. Total: 31 alunos
 n. alunos Turma de terça-feira 14 hs 14 Distribuição de notas 12 10 8 6 4 2 Média = 6,7 0 0 -- 2 2 -- 4 4 -- 6 6 -- 8 8 -- 10 notas 18 alunos Total: 31 alunos BANCO DE DADOS BIOLÓGICOS Aula 12 Estudo dirigido
n. alunos Turma de terça-feira 14 hs 14 Distribuição de notas 12 10 8 6 4 2 Média = 6,7 0 0 -- 2 2 -- 4 4 -- 6 6 -- 8 8 -- 10 notas 18 alunos Total: 31 alunos BANCO DE DADOS BIOLÓGICOS Aula 12 Estudo dirigido
Dados Moleculares x Morfológicos
 Evolução Molecular Dados Moleculares x Morfológicos Hereditários Descrição não ambígua Mais fácil estabelecer homologia Permite comparações de espécies distantes Abundantes Fatores ambientais Diferenças
Evolução Molecular Dados Moleculares x Morfológicos Hereditários Descrição não ambígua Mais fácil estabelecer homologia Permite comparações de espécies distantes Abundantes Fatores ambientais Diferenças
BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011
 BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011 7ª aula teórica 11 Outubro 2010 Proteínas estruturais e funcionais Organização estrutural das proteínas Estrutura e diferentes funções de proteínas
BIOQUÍMICA I 1º ano de Medicina Ensino teórico 2010/2011 7ª aula teórica 11 Outubro 2010 Proteínas estruturais e funcionais Organização estrutural das proteínas Estrutura e diferentes funções de proteínas
Programa Analítico de Disciplina BQI460 Bioinformática
 0 Programa Analítico de Disciplina Departamento de Bioquímica e Biologia Molecular - Centro de Ciências Biológicas e da Saúde Número de créditos: Teóricas Práticas Total Duração em semanas: 15 Carga horária
0 Programa Analítico de Disciplina Departamento de Bioquímica e Biologia Molecular - Centro de Ciências Biológicas e da Saúde Número de créditos: Teóricas Práticas Total Duração em semanas: 15 Carga horária
Mendeley. Maio de 2017.
 Mendeley Maio de 2017. O QUE É o Mendeley e como ele pode me ajudar? O Mendeley é um gerenciador de referências bibliográficas. As principais funções dos gerenciadores de referências são: Criar e organizar
Mendeley Maio de 2017. O QUE É o Mendeley e como ele pode me ajudar? O Mendeley é um gerenciador de referências bibliográficas. As principais funções dos gerenciadores de referências são: Criar e organizar
ANÁLISE DE TANDEM REPEATS CODIFICANTES EM GENOMAS BACTERIANOS
 5ª Jornada Científica e Tecnológica e 2º Simpósio de Pós-Graduação do IFSULDEMINAS 06 a 09 de novembro de 2013, Inconfidentes/MG ANÁLISE DE TANDEM REPEATS CODIFICANTES EM GENOMAS BACTERIANOS Vinícius A.
5ª Jornada Científica e Tecnológica e 2º Simpósio de Pós-Graduação do IFSULDEMINAS 06 a 09 de novembro de 2013, Inconfidentes/MG ANÁLISE DE TANDEM REPEATS CODIFICANTES EM GENOMAS BACTERIANOS Vinícius A.
Programação Dinâmica. Prof. Anderson Almeida Ferreira. Adaptado do material elaborado por Andrea Iabrudi Tavares
 Programação Dinâmica Prof. Anderson Almeida Ferreira Adaptado do material elaborado por Andrea Iabrudi Tavares Programação Dinâmica 1950, Bellman Evitar recálculos dos subproblemas em comum Menor para
Programação Dinâmica Prof. Anderson Almeida Ferreira Adaptado do material elaborado por Andrea Iabrudi Tavares Programação Dinâmica 1950, Bellman Evitar recálculos dos subproblemas em comum Menor para
Profa. Dra. Cecília Dias Flores
 Profa. Dra. Cecília Dias Flores Regente pela Disciplina de Bioinformática - Curso de Biomedicina Depto. Ciências Exatas e Sociais Aplicadas Coordenadora do curso Informática Biomédica PPG em Ciências da
Profa. Dra. Cecília Dias Flores Regente pela Disciplina de Bioinformática - Curso de Biomedicina Depto. Ciências Exatas e Sociais Aplicadas Coordenadora do curso Informática Biomédica PPG em Ciências da
Construção de Filogenias Moleculares
 TP Basic Topic 2 Construção de Filogenias Moleculares Steps to Construct a Molecular Phylogeny Method Choose gene(s) of interest Example YFG-sp1 Identify homologs YFG-sp6 YFG-sp2 YFG-sp1 YFG-sp3 YFG-sp4
TP Basic Topic 2 Construção de Filogenias Moleculares Steps to Construct a Molecular Phylogeny Method Choose gene(s) of interest Example YFG-sp1 Identify homologs YFG-sp6 YFG-sp2 YFG-sp1 YFG-sp3 YFG-sp4
SISTEMÁTICA FILOGENÉTICA. Aula 6: inferência filogenética Parcimônia
 SISTEMÁTICA FILOGENÉTICA Aula 6: inferência filogenética Parcimônia Inferência Qual árvore é a que melhor representa a relação evolutiva entre as espécies? 2 Inferência Um exemplo em Carnivora > Dentição,
SISTEMÁTICA FILOGENÉTICA Aula 6: inferência filogenética Parcimônia Inferência Qual árvore é a que melhor representa a relação evolutiva entre as espécies? 2 Inferência Um exemplo em Carnivora > Dentição,
Bioinformática. Alinhamento de Sequências. Prof. Msc. Rommel Ramos
 Bioinformática Alinhamento de Sequências Prof. Msc. Rommel Ramos 2013 Sumário 1. Comparação de Sequências 2. O que é alinhamento? 3. Tipos de Alinhamento 4. Algoritmos 5. Métodos de Alinhamento Comparação
Bioinformática Alinhamento de Sequências Prof. Msc. Rommel Ramos 2013 Sumário 1. Comparação de Sequências 2. O que é alinhamento? 3. Tipos de Alinhamento 4. Algoritmos 5. Métodos de Alinhamento Comparação
TÍTULO: ANÁLISE DA SEMELHANÇA ESTRUTURAL ENTRE PROTEÍNAS ATRAVÉS DE MÉTODOS MATEMÁTICOS
 Anais do Conic-Semesp. Volume 1, 2013 - Faculdade Anhanguera de Campinas - Unidade 3. ISSN 2357-8904 TÍTULO: ANÁLISE DA SEMELHANÇA ESTRUTURAL ENTRE PROTEÍNAS ATRAVÉS DE MÉTODOS MATEMÁTICOS CATEGORIA: CONCLUÍDO
Anais do Conic-Semesp. Volume 1, 2013 - Faculdade Anhanguera de Campinas - Unidade 3. ISSN 2357-8904 TÍTULO: ANÁLISE DA SEMELHANÇA ESTRUTURAL ENTRE PROTEÍNAS ATRAVÉS DE MÉTODOS MATEMÁTICOS CATEGORIA: CONCLUÍDO
Programação Dinâmica. Prof. Anderson Almeida Ferreira
 Programação Dinâmica Prof. Anderson Almeida Ferreira Programação Dinâmica 1950, Bellman Evitar recálculos dos subproblemas em comum Menor para maior (bottom-up) Tabelas ou memorização É uma técnica de
Programação Dinâmica Prof. Anderson Almeida Ferreira Programação Dinâmica 1950, Bellman Evitar recálculos dos subproblemas em comum Menor para maior (bottom-up) Tabelas ou memorização É uma técnica de
English version at the end of this document
 English version at the end of this document Ano Letivo 2016-17 Unidade Curricular FUNDAMENTOS DE BIOINFORMÁTICA Cursos CIÊNCIAS BIOMÉDICAS (1.º ciclo) Unidade Orgânica Reitoria - Centro de Novos Projectos
English version at the end of this document Ano Letivo 2016-17 Unidade Curricular FUNDAMENTOS DE BIOINFORMÁTICA Cursos CIÊNCIAS BIOMÉDICAS (1.º ciclo) Unidade Orgânica Reitoria - Centro de Novos Projectos
Busca em banco de dados
 Busca em banco de dados Busca em banco de dados A quantidade imensa de dados existentes nos bancos públicos torna critica a existência de ferramentas eficientes que permitam a recuperação de dados desejados
Busca em banco de dados Busca em banco de dados A quantidade imensa de dados existentes nos bancos públicos torna critica a existência de ferramentas eficientes que permitam a recuperação de dados desejados
Bioinformática DCC/FCUP
 Bioinformática DCC/FCUP 2012/2013 Pedro Ribeiro Unidade 3 Alinhamento de Sequências (baseado nos slides de Vítor Costa/DCC-FCUP e Sushmita Roy/UWisconsin) Objectivos desta unidade Homologia Alinhamento
Bioinformática DCC/FCUP 2012/2013 Pedro Ribeiro Unidade 3 Alinhamento de Sequências (baseado nos slides de Vítor Costa/DCC-FCUP e Sushmita Roy/UWisconsin) Objectivos desta unidade Homologia Alinhamento
IBM1029 Introdução à Bioinformática. O Início da Bioinformática 27/03/2017. Aula 2. O Início. Bioionformática: definição
 IBM1029 Introdução à Bioinformática Profa Dra Silvana Giuliatti Departamento de Genética FMRP silvana@fmrp.usp.br O Início da Bioinformática Aula 2 O Início Trabalho de Margaret Dayhoff e colaboradores:
IBM1029 Introdução à Bioinformática Profa Dra Silvana Giuliatti Departamento de Genética FMRP silvana@fmrp.usp.br O Início da Bioinformática Aula 2 O Início Trabalho de Margaret Dayhoff e colaboradores:
Tópicos Especiais em Inteligência Artificial COS746. Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro
 Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Introdução Fundamentos: 1. Algoritmos e Estruturas de Dados (COS) 2. Estatística:
Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Introdução Fundamentos: 1. Algoritmos e Estruturas de Dados (COS) 2. Estatística:
Gene de um organismo Eukariota. Intrões. Codão STOP UTR 5 3. Codão ATG. Exões. Transcrição. 5 Cap 3 poly-a. Splicing. Proteína 3/17/2005 3
 lgoritmos para a Detecção de Promotores otores em Sequências de DN na eresa Freitas INES-ID/IS ID/IS 3/17/5 1 omo analisar todos estes dados? 3/17/5 1 ene de um organismo Eukariota SS Região promotora
lgoritmos para a Detecção de Promotores otores em Sequências de DN na eresa Freitas INES-ID/IS ID/IS 3/17/5 1 omo analisar todos estes dados? 3/17/5 1 ene de um organismo Eukariota SS Região promotora
Prof. João Carlos Setubal
 Prof. João Carlos Setubal QBQ 102 Aula 2 (biomol) Como genes codificam proteínas 2 Genes e proteínas DNA Proteína 3 Hugues Sicotte, NCBI Proteínas são as moléculas trabalhadoras dos organismos nós somos
Prof. João Carlos Setubal QBQ 102 Aula 2 (biomol) Como genes codificam proteínas 2 Genes e proteínas DNA Proteína 3 Hugues Sicotte, NCBI Proteínas são as moléculas trabalhadoras dos organismos nós somos
Evolução Molecular. "Nothing in Biology Makes Sense Except in the Light of Evolution. Theodosius Dobzhansky
 "Nothing in Biology Makes Sense Except in the Light of Evolution Theodosius Dobzhansky Evolução Evolução Evolução Genótipo + Ambiente = Fenótipo Parental F1 F2 Evolução Evolução = mudança (características
"Nothing in Biology Makes Sense Except in the Light of Evolution Theodosius Dobzhansky Evolução Evolução Evolução Genótipo + Ambiente = Fenótipo Parental F1 F2 Evolução Evolução = mudança (características
Busca em banco de dados
 Busca em banco de dados Busca em banco de dados A quantidade imensa de dados existentes nos bancos públicos torna critica a existência de ferramentas eficientes que permitam a recuperação de dados desejados
Busca em banco de dados Busca em banco de dados A quantidade imensa de dados existentes nos bancos públicos torna critica a existência de ferramentas eficientes que permitam a recuperação de dados desejados
Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias
 João de Abreu e Tôrres Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias Dissertação apresentada ao Departamento de Ciência da Computação da UFMG como
João de Abreu e Tôrres Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias Dissertação apresentada ao Departamento de Ciência da Computação da UFMG como
Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias
 João de Abreu e Tôrres Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias Dissertação apresentada ao Departamento de Ciência da Computação da UFMG como
João de Abreu e Tôrres Protein Classification Tool: Uma ferramenta para anotação de proteínas utilizando bases secundárias Dissertação apresentada ao Departamento de Ciência da Computação da UFMG como
ESTATÍSTICAS PARA INFERIR PADRÕES SELECÇÃO
 ESTATÍSTICAS PARA INFERIR PADRÕES DEMOGRÁFICOS E SELECÇÃO Inferências demográficas e selectivas Os fenómenos demográficos (expansão ou redução do efectivo populacional, subdivisão, migração) e selectivos
ESTATÍSTICAS PARA INFERIR PADRÕES DEMOGRÁFICOS E SELECÇÃO Inferências demográficas e selectivas Os fenómenos demográficos (expansão ou redução do efectivo populacional, subdivisão, migração) e selectivos
Modelagem Comparativa de Proteínas
 Modelagem Comparativa de Proteínas Bioinformática Estrutural Aula 3 3 de Junho de 2013 Paula Kuser Falcão Laboratório de Bioinformática Aplicada Embrapa Informática Agropecuária Por que predizer a estrutura?
Modelagem Comparativa de Proteínas Bioinformática Estrutural Aula 3 3 de Junho de 2013 Paula Kuser Falcão Laboratório de Bioinformática Aplicada Embrapa Informática Agropecuária Por que predizer a estrutura?
Análise e Projeto de Algoritmos
 Análise e Projeto de Algoritmos Mestrado em Ciência da Computação Prof. Dr. Aparecido Nilceu Marana Faculdade de Ciências I think the design of efficient algorithms is somehow the core of computer science.
Análise e Projeto de Algoritmos Mestrado em Ciência da Computação Prof. Dr. Aparecido Nilceu Marana Faculdade de Ciências I think the design of efficient algorithms is somehow the core of computer science.
Algoritmo. Exemplo. Definição. Programação de Computadores Comparando Algoritmos. Alan de Freitas
 Algoritmos Programação de Computadores Comparando Algoritmos Um algoritmo é um procedimento de passos para cálculos. Este procedimento é composto de instruções que definem uma função Até o momento, vimos
Algoritmos Programação de Computadores Comparando Algoritmos Um algoritmo é um procedimento de passos para cálculos. Este procedimento é composto de instruções que definem uma função Até o momento, vimos
MIDB-OP: um Modelo de Integração de Dados Biológicos apoiado em Ontologias e Procedência de dados Caroline Beatriz Perlin
 MIDB-OP: um Modelo de Integração de Dados Biológicos apoiado em Ontologias e Procedência de dados Caroline Beatriz Perlin Orientador: Prof. Dr. Ricardo Rodrigues Ciferri Agenda Introdução Bancos de dados
MIDB-OP: um Modelo de Integração de Dados Biológicos apoiado em Ontologias e Procedência de dados Caroline Beatriz Perlin Orientador: Prof. Dr. Ricardo Rodrigues Ciferri Agenda Introdução Bancos de dados
Representação Interação-Transformação para Regressão Simbólica
 Representação Interação-Transformação para Regressão Simbólica Prof. Fabricio Olivetti de França Federal University of ABC Center for Mathematics, Computation and Cognition (CMCC) Heuristics, Analysis
Representação Interação-Transformação para Regressão Simbólica Prof. Fabricio Olivetti de França Federal University of ABC Center for Mathematics, Computation and Cognition (CMCC) Heuristics, Analysis
Banco de Dados Biológicos
 Pontifícia Universidade Católica de Goiás Departamento de Biologia Disciplina: Bioinformática Bio1015 Banco de Dados Biológicos Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com INTRODUÇÃO BANCO
Pontifícia Universidade Católica de Goiás Departamento de Biologia Disciplina: Bioinformática Bio1015 Banco de Dados Biológicos Prof. Macks Wendhell Gonçalves, Msc mackswendhell@gmail.com INTRODUÇÃO BANCO
ALGORITMOS AVANÇADOS UNIDADE I Análise de Algoritmo - Notação O. Luiz Leão
 Luiz Leão luizleao@gmail.com http://www.luizleao.com Conteúdo Programático 1.1 - Algoritmo 1.2 - Estrutura de Dados 1.2.1 - Revisão de Programas em C++ envolvendo Vetores, Matrizes, Ponteiros, Registros
Luiz Leão luizleao@gmail.com http://www.luizleao.com Conteúdo Programático 1.1 - Algoritmo 1.2 - Estrutura de Dados 1.2.1 - Revisão de Programas em C++ envolvendo Vetores, Matrizes, Ponteiros, Registros
Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática
 Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática Hoje iremos trabalhar com dois programas free desenvolvidos pelo Sanger institute: Artemis e ACT. Artemis
Tutorial Introdução a anotação e comparação de genomas Tiago Mendes Doutorando em Bionformática Hoje iremos trabalhar com dois programas free desenvolvidos pelo Sanger institute: Artemis e ACT. Artemis
Algoritmos e Estruturas de Dados I Prof. Tiago Eugenio de Melo
 Algoritmos e Estruturas de Dados I Prof. Tiago Eugenio de Melo tmelo@uea.edu.br www.tiagodemelo.info Observações O conteúdo dessa aula é parcialmente proveniente do Capítulo 11 do livro Fundamentals of
Algoritmos e Estruturas de Dados I Prof. Tiago Eugenio de Melo tmelo@uea.edu.br www.tiagodemelo.info Observações O conteúdo dessa aula é parcialmente proveniente do Capítulo 11 do livro Fundamentals of
DANIELE FERREIRA DA SILVA FILOGENIA MOLECULAR E GENÔMICA COMPARATIVA DE BACTÉRIAS GRAM-POSITIVAS DO TRATO GASTROINTESTINAL
 DANIELE FERREIRA DA SILVA FILOGENIA MOLECULAR E GENÔMICA COMPARATIVA DE BACTÉRIAS GRAM-POSITIVAS DO TRATO GASTROINTESTINAL Tese apresentada à Universidade Federal de Viçosa, como parte das exigências do
DANIELE FERREIRA DA SILVA FILOGENIA MOLECULAR E GENÔMICA COMPARATIVA DE BACTÉRIAS GRAM-POSITIVAS DO TRATO GASTROINTESTINAL Tese apresentada à Universidade Federal de Viçosa, como parte das exigências do
Tópicos Especiais em Inteligência Artificial COS746. Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro
 Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven/C. David Page para BMI/CS 576,
Tópicos Especiais em Inteligência Artificial COS746 Vítor Santos Costa COPPE/Sistemas Universidade Federal do Rio de Janeiro Agradecimento Copiado dos slides de Mark Craven/C. David Page para BMI/CS 576,
IFSC Campus Lages. Tradução. Biologia Molecular Prof. Silmar Primieri
 IFSC Campus Lages Tradução Biologia Molecular Prof. Silmar Primieri Relação DNA RNA Proteína Estrutura das proteínas Gene - Proteína Hipótese Gene - Proteina Os genes são responsáveis pelo funcionamento
IFSC Campus Lages Tradução Biologia Molecular Prof. Silmar Primieri Relação DNA RNA Proteína Estrutura das proteínas Gene - Proteína Hipótese Gene - Proteina Os genes são responsáveis pelo funcionamento
COLÉGIO PEDRO II CAMPUS TIJUCA II. DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI
 COLÉGIO PEDRO II CAMPUS TIJUCA II DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI 1º & 2º TURNOS 3ª SÉRIE / ENSINO MÉDIO REGULAR & INTEGRADO ANO LETIVO 2015 PROFESSORES: FRED & PEDRO
COLÉGIO PEDRO II CAMPUS TIJUCA II DEPARTAMENTO DE BIOLOGIA E CIÊNCIAS COORD.: PROFa. CRISTIANA LIMONGI 1º & 2º TURNOS 3ª SÉRIE / ENSINO MÉDIO REGULAR & INTEGRADO ANO LETIVO 2015 PROFESSORES: FRED & PEDRO
Programa de Pós-Graduação Stricto Sensu em Biologia Computacional e Sistemas. Seleção de Mestrado 2012-A
 Programa de Pós-Graduação Stricto Sensu em Biologia Computacional e Sistemas Seleção de Mestrado 2012-A INSTRUÇÕES (LEIA ATENTAMENTE ANTES DE PREENCHER A PROVA): a. Identifique sua prova unicamente com
Programa de Pós-Graduação Stricto Sensu em Biologia Computacional e Sistemas Seleção de Mestrado 2012-A INSTRUÇÕES (LEIA ATENTAMENTE ANTES DE PREENCHER A PROVA): a. Identifique sua prova unicamente com
Complexidade de algoritmos Notação Big-O
 Complexidade de algoritmos Notação Big-O Prof. Byron Leite Prof. Tiago Massoni Engenharia da Computação Poli - UPE Motivação O projeto de algoritmos é influenciado pelo estudo de seus comportamentos Problema
Complexidade de algoritmos Notação Big-O Prof. Byron Leite Prof. Tiago Massoni Engenharia da Computação Poli - UPE Motivação O projeto de algoritmos é influenciado pelo estudo de seus comportamentos Problema
SISTEMÁTICA - FILOGENIAS LISTA I 10 QUESTÕES PROFESSOR: CHARLES REIS CURSO EXPOENTE
 SISTEMÁTICA - FILOGENIAS LISTA I 10 QUESTÕES PROFESSOR: CHARLES REIS CURSO EXPOENTE 1. (Unicamp/2017) O cladograma abaixo representa relações evolutivas entre membros da Superfamília Hominoidea, onde se
SISTEMÁTICA - FILOGENIAS LISTA I 10 QUESTÕES PROFESSOR: CHARLES REIS CURSO EXPOENTE 1. (Unicamp/2017) O cladograma abaixo representa relações evolutivas entre membros da Superfamília Hominoidea, onde se
Instruções 6 Técnicas
 Instruções 6 Técnicas ISSN Dezembro, 2001 Campinas, SP Entendendo e Interpretando os Parâmetros Utilizados por BLAST Roberto Hiroshi Higa 1 O advento da tecnologia de obtenção rápida de seqüências de DNA,
Instruções 6 Técnicas ISSN Dezembro, 2001 Campinas, SP Entendendo e Interpretando os Parâmetros Utilizados por BLAST Roberto Hiroshi Higa 1 O advento da tecnologia de obtenção rápida de seqüências de DNA,