Consolidado de normas da COGEN
|
|
|
- Ester Conceição Lancastre
- 8 Há anos
- Visualizações:
Transcrição
1 Consolidado de normas da COGEN (Versão BETA 0.1) Agência Nacional de Vigilância Sanitária (ANVISA) Superintendência de Medicamentos e Produtos Biológicos (SUMED) Gerência Geral de Medicamentos (GGMED) Coordenação de Medicamentos Específicos, Notificados e Gases Medicinais (COGEN) Brasília, outubro de
 Coordenação de Medicamentos Específicos, Notificados e Gases")
2 É permitida a reprodução parcial ou total desta obra, desde que citada a fonte. Diretor-Presidente Jarbas Barbosa da Silva Júnior Diretores Renato Alencar Porto José Carlos Magalhães da Silva Moutinho Ivo Bucaresky Superintendência de Medicamentos e Produtos Biológicos Meiruze Souza Freitas Gerência Geral de Medicamentos Ricardo Ferreira Borges Coordenação de Medicamentos Específicos, Notificados e Gases Medicinais João Paulo Silvério Perfeito Equipe técnica Cyro Barbosa Caldeira David Edgard Pietro Gabriella Hamu Giudice Juçara Ribeiro Franca Maíra Ribeiro de Souza Melina Cossote Kumoto Raquel Marcolongo Rogério de Castro Barbosa Sávio Rodrigo de Lima 2

3 INTRODUÇÃO A Coordenação de Medicamentos Específicos, Notificados e Gases Medicinais (COGEN) é uma coordenação da Gerência Geral de Medicamentos (GGMED), da Superintendência de Medicamentos e Produtos Biológicos (SUMED) da Agência Nacional de Vigilância Sanitária (ANVISA). Esta coordenação tem por atribuição emitir documentos circunstanciados e conclusivos em relação ao registro, à renovação e ao pós-registro de medicamentos específicos e à notificação de medicamentos de baixo risco e gases medicinais, conforme legislação vigente. Considerando os diversos assuntos que lhe são pertinentes, a COGEN/GGMED/SUMED elaborou este consolidado de normas na sua primeira versão. Nas páginas iniciais, são citadas as normas mais utilizadas no registro, renovação, pós-registro e notificação dos medicamentos supracitados, à exceção dos gases medicinais, cujos regulamentos encontram-se em revisão. 3

4 Índice MEDICAMENTOS ESPECÍFICOS Introdução Normas sobre Medicamentos Específicos: RDC N 24, DE 14 DE JUNHO DE 2011 Dispõe sobre o registro de medicamentos específicos. RDC Nº 48, DE 6 DE OUTUBRO DE 2009 Dispõe sobre realização de alteração, inclusão, suspensão, reativação, e cancelamento pós-registro de medicamentos e dá outras providências RDC N.º 29, DE 17 DE ABRIL DE 2007 Dispõe sobre as regras referentes ao registro e comercialização para a substituição do sistema de infusão aberto para fechado em Soluções Parenterais de Grande Volume. RDC Nº. 269, DE 22 DE SETEMBRO DE O Regulamento Técnico sobre a Ingestão Diária Recomendada (IDR) de Proteínas, Vitaminas e Minerais. RDC Nº 8, DE 2 DE JANEIRO DE 2001 Aprova o Regulamento Técnico que institui as Boas Práticas de Fabricação do Concentrado Polieletrolíticos para Hemodiálise CPHD RDC Nº 78, DE 11 DE ABRIL DE 2003 Dispõe sobre exigências para a renovação do registro de medicamentos para o tratamento do fígado e que contenham indicação profilática como hepatoprotetor. RDC Nº 05, DE 30 DE JANEIRO DE 2015 Dispõe sobre regra de transição de lágrimas artificiais e ou lubrificantes oculares da categoria de produtos para a saúde para a categoria de medicamentos. PORTARIA N 272, DE 08 DE ABRIL DE 1998 Aprova o Regulamento Técnico para fixar os requisitos mínimos exigidos para a Terapia de Nutrição Parenteral. PORTARIA Nº 40, DE 13 DE JANEIRO DE 1998 Regulamento que estabelece normas para Níveis de Dosagens Diárias de Vitaminas e Minerais em Medicamentos. PORTARIA Nº 108, DE 25 DE JULHO DE
 de Proteínas, Vitaminas e Minerais.")
5 Normatiza a composição de produtos para TRO, de acordo com o conceito de reidratação, manutenção e prevenção em TRO contidas nas normas de controle de doenças diarreicas do Ministério da Saúde. Orientações ao setor regulado sobre medicamentos específicos: Orientações sobre medicamentos à base de água do mar Nota técnica: Comprovação de segurança e eficácia de vitaminas, minerais e aminoácidos Adequação das Soluções Parenterais de Grande Volume SPGV ao sistema fechado. Seminário de Orientação ao Setor Regulado na Área de Medicamentos Guia para a condução de estudos não clínicos de toxicologia e segurança farmacológica necessários ao desenvolvimento de medicamentos Guia para Registro de Novas Associações em Dose Fixa Nota técnica: Reenquadramento de lágrimas artificiais e lubrificantes oculares como medicamentos específicos Nota técnica: Eficácia e segurança de medicamentos específicos Orientação de serviço 01/2014: Registro e pós-registro de medicamentos específicos suplementos vitamínicos e minerais de uso oral Orientação de serviço 03/2014: Esclarecimentos e prorrogação de prazo relacionados à Orientação de serviço 01/2014, que trata do registro e pós-registro de medicamentos específicos suplementos vitamínicos e minerais de uso oral Nota técnica: Mudanças relacionadas ao Insumo Farmacêutico Ativo para Medicamentos Específicos Normas sobre Medicamentos Notificados: RESOLUÇÃO RDC Nº 199, DE 26 DE OUTUBRO DE 2006 Dispõe sobre os medicamentos de notificação simplificada. INSTRUÇÃO NORMATIVA Nº 03, DE 28 DE ABRIL DE 2009 Dispõe sobre a atualização do anexo I da da RDC 199 de 2006 e dá outras providências. (Apresenta a lista padronizada, atualizada, dos medicamentos sujeitos à notificação simplificada) Normas Gerais Aplicadas a Medicamentos Específicos e Notificados RDC Nº 17, DE 16 DE ABRIL DE 2010 Dispõe sobre as Boas práticas de fabricação de medicamentos 5

6 RDC Nº 47, DE 08 DE SETEMBRO DE 2009 NÃO APLICÁVEL A MEDICAMENTOS NOTIFICADOS Estabelece regras para elaboração, harmonização, atualização, publicação e disponibilização de bulas de medicamentos para pacientes e para profissionais de saúde. RDC Nº 71, DE 22 DE DEZEMBRO DE 2009 NÃO APLICÁVEL A MEDICAMENTOS NOTIFICADOS Estabelece regras para a rotulagem de medicamentos RDC Nº 59, DE 10 DE OUTUBRO DE 2014 Dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos. RDC Nº 96, DE 17 DE DEZEMBRO DE 2008 Dispõe sobre a propaganda, publicidade, informação e outras práticas cujo objetivo seja a divulgação ou promoção comercial de medicamentos. RE Nº 1, DE 29 DE JULHO DE 2005 Guia para a realização de estudos de estabilidade RE Nº 899, DE 29 DE MAIO DE 2003 Guia para validação de métodos analíticos e bioanalíticos RDC Nº 37, DE 06 DE JULHO DE 2009 Trata da admissibilidade de Farmacopeias estrangeiras LEI Nº 6.360, DE 23 DE SETEMBRO DE 1976 Dispõe sobre a Vigilância Sanitária a que ficam sujeitos os Medicamentos, as Drogas, os Insumos Farmacêuticos e Correlatos, Cosméticos, Saneantes e Outros Produtos, e dá outras Providências RDC Nº 138, DE 29 DE MAIO DE 2003 Dispõe sobre o enquadramento na categoria de venda de medicamentos RDC Nº 305 DE 14 DE NOVEMBRO DE 2002 Ficam proibidos, em todo o território nacional, enquanto persistirem as condições que configurem risco à saúde, o ingresso e a comercialização de matéria-prima e produtos acabados, semielaborados ou a granel para uso em seres humanos, cujo material de partida seja obtido a partir de tecidos/fluidos de animais ruminantes, relacionados às classes de medicamentos, cosméticos e produtos para a saúde, conforme discriminado. RDC 68 DE 28 DE MARÇO DE 2003 Estabelece condições para importação, comercialização, exposição ao consumo dos produtos incluídos na Resolução da Diretoria Colegiada - RDC nº 305, de 14 de novembro de RDC Nº 1.548, DE 23 DE SETEMBRO DE

7 Dispõe sobre a publicação das "Categorias de risco de fármacos destinados às mulheres grávidas" RE Nº 572, DE 05 DE ABRIL DE 2002 Estabelece que os medicamentos contendo o excipiente corante TARTRAZINA (Amarelo FD&C Nº5) devem conter na bula e no cartucho (embalagem externa), de forma claramente visível e destacada, o seguinte aviso: Este produto contém o corante amarelo de TARTRAZINA que pode causar reações de natureza alérgica, entre as quais asma brônquica, especialmente em pessoas alérgicas ao Ácido Acetil Salicílico. RDC Nº 25, DE 29 DE MARÇO DE 2007 Dispõe sobre terceirização de controle de qualidade RDC Nº 04, DE 10 DE FEVEREIRO DE 2009 Dispõe sobre as normas de farmacovigilância para os detentores de registro de medicamentos de uso humano RDC N 09, DE 20 DE FEVEREIRO DE 2015 Dispõe sobre o Regulamento para a realização de ensaios clínicos com medicamentos no Brasil. 7

8 MEDICAMENTOS ESPECÍFICOS Os medicamentos específicos não possuem uma característica universal que os una e que os defina, por essa razão, o enquadramento de um medicamento como específico é muitas vezes complexo. De acordo com a RDC n 24/2011, o medicamento específico é aquele que não pode ser enquadrado nas categorias de medicamento novo, similar, genérico, biológico, fitoterápico ou notificado e, ainda, cujo ativo não seja passível de ensaio de bioequivalência frente a um medicamento comparador. Deste modo, devido à amplitude dessa definição, a legislação vigente traz uma lista das categorias de produtos que são classificados como medicamentos específicos.contudo, há possibilidade de enquadramento como medicamento específico de casos não listados no Art. 5º, mas que atendam ao Art. 3º da RDC nº. 24/2011, sendo o enquadramento destes produtos avaliado caso a caso. Por vezes, os medicamentos específicos podem ser confundidos com outras categorias de produtos, como produtos para saúde, alimentos ou cosméticos. Para a diferenciação, a premissa básica é que os específicos são medicamentos e, por conseguinte, devem obrigatoriamente ter finalidade profilática, curativa, paliativa ou para fins de diagnóstico. 1. REGISTRO DE MEDICAMENTOS ESPECÍFICOS 1.1 Informações gerais O registro de medicamentos específicos segue os demais regulamentos para medicamentos a serem registrados na ANVISA, ou seja, Lei n /1976 e Decreto n /2013, como normas orientadoras gerais para registro de medicamentos; regras para bulas de medicamentos: RDC n. 47/2009; regras para embalagens e nome comercial: RDC n. 71/2009 e RDC nº. 59/2014; regras para publicidade de medicamentos: RDC n. 96/2008; Guia para realização de estudos de estabilidade de medicamentos : RE n. 01/2005; e o Guia para validação de metodologias analíticas e bioanalíticas de medicamentos : RE n. 899/2003. Além dos requisitos gerais que constam na RDC n. 24/2011, algumas categorias de medicamento específico possuem legislação complementar. Para as Soluções Parenterais de Grande Volume, o registro deve estar conforme o disposto na RDC n. 29/2007 que trata dos requisitos para comprovação de produção em sistema fechado de infusão intravenosa. Para os Concentrados Polieletrolíticos para Hemodiálise (CPHD) devem ser seguidos os critérios de qualidade, segurança e eficácia descritos na RDC n. 08/2001, enquanto para nutrição parenteral, os critérios a serem seguidos estão descritos na Portaria n. 272/1998. Na Portaria n. 108/1991, constam as fórmulas, indicações e critérios de qualidade a serem seguidos para os medicamentos de Terapia de Reidratação Oral. No caso das vitaminas e minerais de uso oral, quando ao menos um princípio ativo estiver acima de 100% da Ingestão Diária Recomendada (IDR) descrita na RDC n. 269/2005, o produto deve ser registrado como medicamento. Além disso, a restrição de venda do medicamento assim como os limites de segurança estão descritos na Portaria n. 40/1998. Para os aminoácidos, atualmente não há na legislação sanitária brasileira limites estabelecidos de IDR e de segurança, assim são aceitos os limites estabelecidos em códigos oficiais de outros países. 1.2 Documentação O dossiê de registro é composto por uma parte legal e outra técnica. Na parte legal, é exigida a apresentação dos seguintes documentos atualizados: Cópia do protocolo da notificação da produção de lotes piloto; Cópia de licença de funcionamento da empresa (Alvará Sanitário); Certificado de Responsabilidade Técnica (CRT) emitido pelo Conselho Regional de Farmácia (CRF); cópia do Certificado de Boa Práticas de Fabricação e Controle (CBPFC) 8

9 emitido pela Anvisa para a linha de produção na qual o medicamento será fabricado e formulários de petição (FP1 e FP2) preenchidos, contendo todas as informações sobre a composição do produto (matéria(s)-prima(s) ativa(s) e excipiente(s)), nome comercial, forma farmacêutica, embalagens, restrição de venda, prazo de validade e cuidados de conservação). Na parte técnica, por sua vez, é exigida a apresentação dos seguintes documentos: relatórios de estabilidade, produção e controle de qualidade; dados do derivado vegetal, do fitofármaco, do derivado de síntese ou semi-síntese e do opoterápico (quando presentes); modelos de bula, rótulo e embalagem; documentação referente a cada local de fabricação (caso a empresa solicite o registro em mais de um local) e relatório de segurança e eficácia (quando aplicável). 1.3 Estabilidade O estudo de estabilidade deve ser projetado para verificação das características físicas, químicas, biológicas e microbiológicas de um produto farmacêutico no prazo de validade esperado. Os resultados são usados para estabelecer ou confirmar esse prazo de validade e recomendar as condições de armazenamento. A RE n. 01/2005, além de outras informações, preconiza três tipos de estudos para fins de registro e renovação de registro de medicamentos na Anvisa: o acelerado, o de longa duração e o de acompanhamento. Nestes três, os medicamentos devem estar acondicionados em suas embalagens primárias. O estudo de estabilidade acelerado objetiva acelerar a degração química e as mudanças físicas de um medicamento, a fim de predizer o seu prazo de validade, além de avaliar o impacto de curtas exposições a condições fora daquelas estabelecidas no rótulo (que podem ocorrer, eventualmente, durante o transporte). Já o estudo de longa duração tem como objetivo verificar as características físicas, químicas, biológicas e microbiológicas do medicamento e estabelecer ou confirmar o seu prazo de validade, além de recomendar suas condições de armazenamento. Os resultados finais do estudo de longa duração, assim como a declaração do prazo de validade e cuidados definitivos de conservação, devem ser apresentados à Anvisa logo que concluídos, na forma de aditamento ao processo. Por fim, o estudo de estabilidade de acompanhamento deve ser realizado para verificar se o medicamento mantém as características demonstradas no estudo de longa duração. Quando a empresa produzir mais de 15 lotes do medicamento, por ano, ela deverá conduzir o estudo de estabilidade de acompanhamento com 1 lote anual do produto. Quando a empresa produzir uma quantidade inferior a 15 lotes por ano, o estudo deverá ser conduzido com um lote do produto a cada 2 anos. Esse estudo deve ser disponibilizado no momento da inspeção na indústria farmacêutica e enviado na petição de renovação de registro. No momento do peticionamento do registro, devem ser apresentados os relatórios do estudo de estabilidade acelerado completo e do estudo de longa duração em andamento, com os resultados de todos os testes realizados conforme preconizado pela legislação vigente. Testes adicionais podem ser necessários para garantir a segurança e eficácia do medicamento. Essa avaliação crítica deve ser feita pela empresa no momento do desenvolvimento do medicamento. Para alguns medicamentos, a RDC n. 24/2011 solicita informações sobre o fármaco que são importantes e de grande impacto para o produto. Assim, devido às peculiaridades de alguns princípios ativos, sua rota de síntese, seus contaminantes e outras informações devem ser fornecidas no momento do registro. Do mesmo modo, o estudo de estabilidade deve ser conduzido levando-se em consideração esses fatores. Nestes casos, para garantir a qualidade, a segurança e a avaliação da reprodutibilidade dos resultados, deve-se produzir lotes-pilotos e conduzir os estudos de estabilidade com amostras de tamanho adequado para cada fabricante de fármaco. 9
; modelos de bula, rótulo e embalagem; documentação referente a cada local de fabricação (caso a empresa solicite o registro em mais de")
10 1.3.1 Necessidade de doseamento de ftalato nos estudos de estabilidade das embalagens de PVC. Os ftalatos compreendem o principal grupo de plastificantes monoméricos empregados na produção de PVC (BUSZARD, 1984). Dentre estes polímeros, o ftalato de di-(2-etilexila) (DEHP) é o plastificante mais utilizado, abrangendo 50% de todos os plastificantes ftalatos (WHO, 1992). Estima-se que, no mínimo 95% do DEHP produzido seja utilizado como plastificante, particularmente em produtos de PVC (EPA, 1996; WHO, 1992). Conforme enuncia a Farmacopeia Britânica: os polímeros empregados na produção de embalagens não devem incluir em sua composição qualquer substância que possa ser extraída pelo conteúdo da embalagem, de forma a prevenir alterações na eficácia ou estabilidade das soluções nelas contidas ou apresentar risco de toxicidade aos pacientes e consumidores. A Farmacopeia Europeia define em 40% o quantitativo máximo de ftalato de di-(2- etilexila) (DEHP) presente nas embalagens de PVC para uso em soluções aquosas pra infusão intravenosa. Outros aditivos e suas concentrações máximas permitidas também estão descritas na Farmacopeia Europeia. Devido a sua estrutura e a sua lipossolubilidade, os ftalatos podem ser absorvidos facilmente pelo organismo, podendo produzir efeitos adversos à saúde humana. Assim, o principal plastificante desta classe, o DEHP, tem sido objeto de inúmeros estudos toxicológicos (WHO, 1992) Os resultados dos diversos estudos toxicológicos serviram de embasamento para a publicação de várias revisões bibliográficas sobre o DEHP: Di(2-ethylhexyl)phthalate (DEHP): human metabolism and internal exposure- - an update and latest results de Koch e col. (2006); Urinary phthalate metabolites and semen quality: a review of a potential biomarker of susceptibility de Hauser (2008); Plasticizers, infant nutrition and reproductive health de Latini e col. (2004) entre outras. Estudos relatam a ocorrência de hiperplasia e hipertrofia hepática e a proliferação de peroxissomos em ratos e camundongos após administração de ftalato. Doses elevadas do produto acarretaram em aumento na incidência de tumores hepáticos nos animais. Diversos animais apresentaram atrofia testicular após a exposição ao ftalato (WHO, 1992). Ademais, diversos estudos também relataram alterações hepatotóxicas após a administração do ftalato (BARBER et al., 1987; EAGON et al., 1994), (YANAGITA et al., 1987), (WINBERG & BADR, 1995). Poon e colaboradores (1997) descreveram alterações histológicas na tireoide após exposição ao ftalato. A Organização Mundial de Saúde (OMS) recomenda que dispositivos médicos e produtos que possam contribuir para a introdução de DEHP no organismo sejam estudados visando reduzir a exposição ao DEHP por via intravenosa (IV) (WHO, 1992). Assim, seguindo o entendimento do item 2.9 da RE 01/2005 todo relatório de estudo de estabilidade deve apresentar quantificação de produtos de degradação. Entende-se que o ftalato, apesar de não ser um produto de degradação direto do princípio ativo, é tóxico e lixiviavel dos plásticos PVC e por isso deve ser dosado nos estudos de estabilidade. Ademais, alguns pesquisadores - Smistad e col. (1989) e Arbin & Östelius (1980) - detectaram DEHP em soluções de NaCl 0.9% e de glicose 5% contidas em bolsas flexíveis de PVC. Esses achados reforçam ainda mais a necessidade de se realizar o doseamento de ftalatos durante os estudos de estabilidade. 1.4 Terceirização A empresa pode optar por terceirizar uma parte da produção ou do controle de qualidade do medicamento, mas deverá selecionar laboratórios habilitados pela Rede Brasileira de Laboratórios em Saúde (REBLAS) ou outras empresas fabricantes de medicamentos que tenham Certificado de Boas Práticas de Fabricação e Controle (CBPFC) atualizado. Informações sobre contratos de terceirização podem ser obtidas na RDC n. 25/
.")
11 1.5 Controle de qualidade O Controle de qualidade dos medicamentos específicos deve atender a requisistos específicos e às monografias farmacopeicas, caso existam. Nos casos em que não houver monografias farmacopeicas, deve-se realizar uma avaliação crítica dos princípios ativos e do produto acabado, a fim de se propor testes que comprovem a manutenção da qualidade e a segurança do medicamento. Deve-se realizar todos os testes requeridos para a forma farmacêutica e incluir, ainda, aqueles que forem considerados importantes considerando as características específicas do produto desenvolvido. Para os medicamentos específicos que possuem teste de dissolução descrito em monografia farmacopeica, este teste deve ser executado. Do mesmo modo, as impurezas descritas em farmacopeia devem ser quantificadas no controle de qualidade e nos estudos de estabilidade. 1.6 Validação As metodologias analíticas que não constem nas farmacopeias reconhecidas pela Anvisa (dipostas na RDC n 37/2009) devem ser validadas seguindo o disposto no Guia para validação de metodologias analíticas e bioanalíticas de medicamentos, publicado por meio da RE n. 899/2003. As metodologias analíticas compendiais, por sua vez, não requerem validação, entretanto, antes de sua implementação, devem existir evidências documentadas de sua adequabilidade às condições operacionais do laboratório, conforme preconiza a RDC n. 17/2010. A escolha dos parâmetros que deverão ser avaliados durante a verificação de métodos compendiais depende da natureza da metodologia avaliada, bem como da complexidade do material ao qual esta será aplicada. Recomenda-se a avaliação, no mínimo, dos parâmetros seletividade, precisão e, no caso de métodos analíticos utilizados para determinação de impurezas, dos limites de detecção e de quantificação. O processo de verificação deverá incluir, ainda, a avaliação do efeito da matriz da amostra nos resultados experimentais, bem como da adequabilidade dos sistemas cromatográficos e das respostas instrumentais. Para medicamentos específicos que contenham derivados vegetais, se o derivado utilizado no produto final tiver sido extraído da planta com o mesmo solvente utilizado na metodologia farmacopeica de monografia da droga vegetal e não houver adição de excipientes, não é necessária a apresentação da validação completa da metodologia analítica, mas é necessária a verificação do método. É necessário que a monografia se refira à apresentação/etapa do produto para a qual se apresentem os testes, ou seja, uma monografia de planta medicinal pode embasar os testes referentes à droga vegetal, mas não ao produto final. Para o produto final, devem ser apresentadas apenas monografias que descrevam o produto conforme solicitado no registro. A validação da metodologia analítica consiste na demonstração de que esta é adequada à finalidade pretendida, qual seja, a determinação qualitativa, quantitativa ou semi-quantitativa do ativo ou dos marcadores (em caso de medicamentos específicos que contenham derivados vegetais). O processo de validação deve envolver a avaliação dos parâmetros: especificidade, linearidade, exatidão, precisão, repetibilidade e robustez. Todas as metodologias utilizadas no controle da qualidade devem ser referenciadas com a indicação da fonte bibliográfica ou de desenvolvimento. De acordo com a RE n. 899/2003, na validação de metodologia analítica, deve-se utilizar substâncias de referência oficializadas pela Farmacopéia Brasileira ou, na ausência destas, por outros códigos autorizados pela legislação vigente. No caso da inexistência dessas substâncias, será admitido o uso de padrões de trabalho, desde que esses sejam devidamente caracterizados. Para medicamentos contendo derivados vegetais, além de substâncias químicas de referência (SQR) dos marcadores da espécie vegetal, também podem ser utilizados os padrões 11

12 oficializados dos extratos (por exemplo, aqueles disponibilizados pela USP), desde que acompanhados de laudo de análise e do perfil cromatográfico do fornecedor. Substâncias isoladas da espécie vegetal ou obtidas de fornecedores qualificados podem ser utilizadas como SQR, desde que acompanhadas de relatório de caracterização completo, incluindo elucidação estrutural inequívoca e determinação de pureza por balanço de massas. Padrões de trabalho dos derivados vegetais podem ser obtidos mediante quantificação de todos os marcadores frente às respectivas SQR e avaliação adequada do perfil cromatográfico. O prazo de validade do extrato qualificado deve ser determinado. 1.7 Comprovação de segurança e eficácia de medicamentos específicos De acordo com a Seção VII da RDC n 24/2011, os medicamentos específicos podem ser isentos de comprovação de segurança ou eficácia. Caso não o sejam, devem comprová-las por meio de dados de literatura técnico-científica, pela apresentação de estudos pré-clínicos e clínicos ou por tradicionalidade de uso, nos termos da norma supracitada. Os medicamentos específicos isentos de comprovação de segurança e eficácia são: medicamentos à base de associações entre vitaminas e/ou minerais e o derivado vegetal de Panax ginseng, com padronização de marcador e posologia diária definida pela Lista de Medicamentos Fitoterápicos e Produtos Tradicionais Fitoterápicos de Registro Simplificado, publicada pela IN nº 02/2014, e cujos níveis de dosagem diária para vitaminas e minerais estejam abaixo dos definidos pela Portaria nº 40/1998, com indicação terapêutica para a prevenção e a recuperação em casos de fadiga física e psíquica, atuando como adaptógeno, e suplementação vitamínico-mineral; medicamentos à base de própolis de uso tópico com as indicações de uso como anti-inflamatório, antisséptico e cicatrizante; medicamentos à base de vitaminas e/ou minerais e/ou aminoácidos, isolados ou associados entre si, de uso oral classificados como medicamentos isentos de prescrição médica; produtos para a prevenção da desidratação e para a manutenção da hidratação oral conforme Portaria nº 108/1991; e CPHD conforme regido pela RDC nº. 08/2001. Todos os outros medicamenteos específicos devem comprovar eficácia e segurança. Para a comprovação por tradicionalidade de uso, deverão ser apresentadas publicações técnico-científicas que serão avaliadas conforme os seguintes critérios: indicação de uso episódico ou para curtos períodos de tempo; indicação para doenças de baixa gravidade ou relacionada à melhoria ou manutenção da saúde; coerência das indicações terapêuticas propostas com as comprovadas pelo uso tradicional; ausência de grupos ou substâncias químicas tóxicas, ou presentes dentro de limites comprovadamente seguros; comprovação de continuidade de uso seguro por período igual ou superior a 10 anos no Brasil; e racionalidade das associações de ativos. Todos esses critérios devem estar comprovados nas publicações técnico-científicas apresentadas para que o medicamento seja considerado seguro e eficaz por tradicionalidade de uso. Além disso, não são permitidas alterações das seguintes características do medicamento durante o período de comercialização igual ou superior a 10 anos: substância(s) ativa(s), qualitativa ou quantitativa; forma farmacêutica, incluindo sistema de liberação; posologia e indicação terapêutica. Nos casos em que for necessário apresentar literatura técnico-científica indexada, só serão aceitos estudos com o(s) princípio(s) ativo(s) e produto com mesma concentração, posologia, forma farmacêutica, via de administração e indicação do produto objeto do registro. Cabe ressaltar que os excipientes podem impactar na farmacocinética e consequentemente na segurança e eficácia do medicamento e estes devem ser levados em 12

13 consideração no levantamento bibliográfico a ser realizado. Além disso, a metodologia do estudo (desfechos, amostragem, randomização, cegamento, comparador e estatística) deve ser avaliada previamente ao peticionamento do registro. Recomenda-se que sejam utilizados como referência estudos com boa qualidade metodológica e bom nível de evidência científica e que estes estudos permitam extrapolar os achados para o medicamento objeto de registro, bem como para a população alvo. A avaliação crítica dos estudos é fundamental para minimizar o risco quanto à segurança e à eficácia do medicamento. Desta forma, as revisões não-sistemáticas podem ser utilizadas como referência no desenvolvimento do produto, contudo, somente as revisões sistemáticas serão aceitas para compor o relatório técnico-científico. Por fim, somente estudos apresentados na íntegra serão considerados e a apresentação apenas dos resumos não serão consideradas. No caso de associações em doses fixas para uso oral, o Guia para Registro de Novas Associações em Doses Fixas traz orientações sobre os requisitos regulatórios de eficácia e segurança. Nos casos em que a legislação preconiza a realização de estudos clínicos e não clínicos para a comprovação de segurança e eficácia, a empresa deve seguir normas específicas para a condução destes estudos. Para estudos não clínicos, o Guia para a Condução de Estudos Não Clínicos de Segurança necessários ao desenvolvimento de Medicamentos traz orientações para a condução de estudos de toxicidade, genotoxicidade, tolerância e carcinogenicidade, dentre outros. Já para os estudos clínicos, o protocolo deve ser previamente anuído pela Anvisa, conforme estabelecido na RDC nº. 09/2015, e a empresa deve seguir as Boas Práticas Clínicas. 1.8 Bulas Todos os medicamentos específicos devem apresentar bula para o paciente e para o profissional de saúde em conformidade com a RDC n. 47/ Rotulagem Os rótulos e embalagens dos medicamentos específicos devem ser elaborados em conformidade com o disposto na RDC nº 71/2009. O nome do medicamento, por sua vez, deve seguir as orientações da RDC n. 59/2014, que dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos.devese observar, também, o nome escolhido para o medicamento, de modo a não induzir o consumidor a erro, ao solicitar um nome semelhante a outro existente no mercado, conforme preconiza a Lei 6.360/ Restrição de venda A restrição de venda para medicamento específico é definida em duas resoluções: RDC n. 138/2003, e Portaria n. 40/1998. A primeira premissa para um medicamento à base de vitaminas, minerais ou aminoácidos ser considerado isento de prescrição é que este deve ter posologia diária máxima abaixo do estabelecido pela Portaria n. 40/1998. Porém, nos casos em que não há limites estabelecidos nessa Portaria, a RDC n. 24/2011, em seu Art. 6º, parágrafo único, estabelece que podem ser aplicados níveis máximos de segurança estabelecidos em outros países, desde que tenham sido regulamentados por meio de códigos oficiais. Com relação à segunda premissa, o medicamento deve ter indicação prevista pela RDC n. 138/2003, devendo ser enquadrado na categoria correta e seguir conforme Art. 1º desta legislação: Todos os medicamentos cujos grupos terapêuticos e indicações terapêuticas estão descritos no Anexo: Lista de Grupos e Indicações Terapêuticas Especificadas (GITE), respeitadas as restrições textuais e de outras normas legais e regulamentares 13

14 pertinentes, são de venda sem prescrição médica, à exceção daqueles administrados por via parenteral que são de venda sob prescrição médica. 2. MEDICAMENTOS DE NOTIFICAÇÃO SIMPLIFICADA A notificação simplificada consiste na comunicação à autoridade sanitária federal da fabricação, importação e comercialização de medicamentos de baixo risco à saúde quando observadas todas as características de uso e qualidade descritas na RDC nº. 199/2006. É processada mediante peticionamento eletrônico, isento de taxa, no sítio eletrônico da Anvisa e não exime as empresas das obrigações do cumprimento das Boas Práticas de Fabricação e Controle e das demais regulamentações sanitárias. Os medicamentos sujeitos à notificação são isentos de prescrição médica, conforme 2º Art. 3º da RDC nº. 199/2006. A primeira etapa da notificação consiste na habilitação da empresa. A habilitação poderá ser solicitada mediante apresentação do Certificado Boas Práticas de Fabricação e Controle (CBPFC) publicado ou do protocolo de solicitação do pedido de CBPFC, desde que a empresa possua status satisfatório no Banco de dados de inspeções da Anvisa. Somente após a validação pela Anvisa a empresa conseguirá notificar medicamentos. A notificação é realizada apenas por meio eletrônico. Não há necessidade de entrega de documentos físicos. A empresa só poderá notificar os medicamentos constantes no Anexo I da IN nº. 03/2009, que atualizou a Lista padronizada de medicamentos sujeitos à notificação simplificada,conforme as informações nele listadas. Atualmente, com a publicação da IN nº. 03/2009, há 75 produtos passíveis de notificação. 2.1 Controle de qualidade As especificações analíticas do medicamento devem estar de acordo com a monografia inscrita em compêndio oficialmente reconhecido pela Anvisa, de acordo com a RDC n. 37/2009. Na ausência de monografia oficial para o produto acabado, deverão ser realizados os testes descritos nos métodos gerais da edição mais recente da Farmacopeia Brasileira, além de outros testes necessários, desenvolvidos pelo fabricante, conforme as características do produto, a fim de garantir a qualidade do medicamento. Todo laudo de análise de controle de qualidade do produto acabado, independente da forma farmacêutica, deve apresentar, no mínimo, as seguintes informações ou justificativa técnica de ausência: características organolépticas/aparência; identificação e teor do(s) princípio(s) ativo(s); limites microbianos: contagem de bactérias e fungos totais e pesquisa de patógenos. Adicionalmente, para as formas farmacêuticas sólidas, a empresa deve apresentar as seguintes informações ou justificativa técnica de ausência: desintegração; dissolução; dureza; peso médio; e umidade. Já para as formas farmacêuticas líquidas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: ph; densidade; viscosidade; e volume ou peso médio. 2.2 Bula e rotulagem A rotulagem dos medicamentos de notificação simplificada deve seguir o estabelecido no Anexo II da RDC nº. 199/2006, sendo dispensada a apresentação de texto bula quando todas as informações preconizadas no Anexo I da IN n 03/2009 estiverem presentes na rotulagem. Caso não seja possível incluir todas as informações na rotulagem, os modelos de bula devem seguir o disposto na RDC n 47/2009. Os medicamentos de notificação simplificada devem citar, em sua rotulagem, seu enquadramento, adotando a frase: MEDICAMENTO DE 14

15 NOTIFICAÇÃO SIMPLIFICADA RDC nº. 199/2006. AFE nº..... Os medicamentos devem adotar para sua identificação, o nome do produto ou sinônimo presentes no Anexo I da IN nº. 03/2009, sendo facultada a adoção de marca ou nome comercial. Desta forma, os medicamentos só podem ser comercializados com o nome do produto ou sinônimo, ou ainda, com o nome comercial, exatamente como informado no momento da notificação. 2.3 Estabilidade Para os ensaios de estabilidade deverão ser seguidas as orientações do Guia para a Realização de Estudos de Estabilidade de medicamentos (RE nº. 01/2005), da mesma maneira que foi orientado para os medicamentos específicos sujeitos ao registro (item 1.3). Ao notificar seus produtos, a empresa deve encaminhar eletronicamente, em um arquivo no formato pdf, os estudos de estabilidade de longa duração, caso já estejam concluídos, ou acelerado acompanhado do de longa duração em andamento, conforme disposto na RE n. 01/2005. Neste último caso, quando concluído o estudo de longa duração, a empresa deverá fazer nova notificação à ANVISA, incluindo o resultado do estudo de longa duração concluído, e proceder com o cancelamento da notificação anterior. O estudo de estabilidade de acompanhamento deve ser realizado para verificar se o medicamento mantém as características demonstradas no estudo de longa duração. 2.4 Terceirização A empresa pode optar por terceirizar uma parte da produção, mas deverá fazê-lo em empresas fabricantes de medicamentos que tenham CBPFC atualizado e que também estejam habilitadas eletronicamente para a notificação simplificada. Informações sobre contratos de terceirização podem ser consultadas na RDC n. 25/ Pós-registro e renovação Tendo em vista que não há previsão petição de alteração pós-registro para medicamentos de notificação simplificada, qualquer alteração de formulação ou de processo produtivo (aumento de tamanho de lote, alteração de excipiente, etc.) de um medicamento de notificação simplificada implicará na realização de novos estudos de estabilidade, cancelamento da notificação anterior e renotificação do medicamento com a apresentação dos novos estudos de estabilidade. As alterações de controle de qualidade podem ser implementadas imediatamente, sem avaliação prévia, desde que atendam ao disposto na IN nº 03/2009. Cabe esclarescer que, conforme Art. 524 da RDC 17/2010, As mudanças devem ser formalmente requisitadas, documentadas e aprovadas antes da implementação. Os registros devem ser mantidos. Deste modo, todas as alterações referentes aos medicamentos de notificação simplificada devem estar registradas nos documentos de controle de mudança da empresa. Todas as notificações devem ser renovadas. O prazo de validade da notificação é de cinco anos e a renovação ocorre mediante nova notificação de cada produto. 2.6 Solicitações de inclusões, alterações e exclusões Solicitações de inclusões, alterações e exclusões de medicamentos no Anexo I da IN nº. 03/2009 poderão ser requeridas por meio do preenchimento do formulário - Anexo III da RDC nº. 199/2006, que deverá ser enviado para 15

16 o juntamente com cópia de toda a referência utilizada para padronização das informações relacionadas ao medicamento. 16

17 RDC N 24, DE 14 DE JUNHO DE 2011 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto nº , de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº. 354 da ANVISA, de 11 de agosto de 2006, republicada no Diário Oficial da União (DOU) de 21 de agosto de 2006, em reunião realizada em 7 de junho de adota a seguinte Resolução da Diretoria Colegiada (RDC) e eu, Diretor- Presidente, determino a sua publicação: Art. 1º Fica aprovado o regulamento técnico que estabelece os requisitos para o registro e a renovação de registro de medicamentos específicos, nos termos desta Resolução. Seção I Objetivo CAPÍTULO I DAS DISPOSIÇÕES INICIAIS Art. 2º Esta Resolução possui o objetivo de definir a categoria de medicamentos específicos e estabelecer os requisitos mínimos para seu registro e renovação de registro. Seção II Abrangência Art. 3º Esta Resolução se aplica aos produtos que se enquadram na categoria de medicamentos específicos. 1º São considerados medicamentos específicos os produtos farmacêuticos, tecnicamente obtidos ou elaborados, com finalidade profilática, curativa ou paliativa não enquadrados nas categorias de medicamento novo, genérico, similar, biológico, fitoterápico ou notificado e cuja (s) substância (s) ativa (s), independente da natureza ou origem, não é passível de ensaio de bioequivalência, frente a um produto comparador. 2º As empresas interessadas no registro de medicamentos específicos deverão cumprir na íntegra os dispositivos desta Resolução e demais normas complementares. 17
 de 21 de agosto de 2006, em reunião realizada em 7 de junho de 2011.")
18 3º Esta Resolução não se aplica aos suplementos vitamínicos e minerais, outros alimentos contendo novos ingredientes, substâncias bioativas, suplementos hidroeletrolíticos para atletas, além de alimentos com alegações de propriedades funcionais, entre outros, que não apresentam finalidade terapêutica ou medicinal. Seção III Definições Art. 4º Para efeito desta Resolução são adotadas as seguintes definições: I - ácidos graxos ômega-3: óleo de ácidos graxos de cadeia longa purificados obtidos a partir de peixes como aqueles das famílias: Ammodytidae, Carangidae, Clupeidae, Engraulidae, Osmeridae, Salmonídeos, Scrombidae e Gadidae que contém ácidos graxos ômega-3, principalmente os ácidos eicosapentaenóico (EPA) e docosahexaenóico (DHA), naturalmente presentes em organismos marinhos; (DHA), ácidos) presentes II - aminoácidos: classe de moléculas orgânicas que estão diretamente relacionadas à síntese protéica, sendo as seguintes substâncias, assim, ordenadas: glicina, alanina, valina, leucina, isoleucina, fenilalanina, asparagina, glutamina, triptofana, prolina, serina, treonina, tirosina, hidroxiprolina, cisteína, cistina, metionina, ácido aspárgico, ácido glutâmico, lisina, arginina e histidina; III - antiácidos: substâncias que atuam contra azia, desconforto estomacal, dor de estômago, dispepsia ou neutralizam a acidez do trato gastrointestinal; IV - Concentrado Polieletrolítico para Hemodiálise (CPHD): concentrado de eletrólitos, com ou sem glicose, apresentado na forma sólida ou líquida para emprego na terapia de diálise renal, após diluição recomendada pelo fabricante e utilizando equipamento específico; V - Certificado de Responsabilidade Técnica (CRT): certificado emitido pelo Conselho Regional de Farmácia que ateste a existência de profissional farmacêutico responsável pela atividade desenvolvida; VI - derivado vegetal: produto da extração da planta medicinal in natura ou da droga vegetal, podendo ocorrer na forma de extrato, tintura, alcoolatura, óleo fixo e volátil, cera, exsudato e outros; VII - doença de baixa gravidade: doença auto-limitante, de evolução benigna, que pode ser tratada sem acompanhamento médico; VIII - droga vegetal: planta medicinal, ou suas partes, que contenham as substâncias, ou classes de substâncias, responsáveis pela ação terapêutica, após processos de coleta, estabilização, quando aplicável, e secagem, podendo estar na forma íntegra, rasurada, triturada ou pulverizada; IX - excipiente: substância adicionada ao medicamento com a finalidade de prevenir alterações, corrigir e/ou melhorar as características organolépticas, biofarmacotécnicas e tecnológicas do medicamento; X - fitofármaco: substância purificada e isolada a partir de matéria-prima vegetal com estrutura química definida e atividade farmacológica. É utilizada como ativo em medicamentos com propriedade profilática, paliativa ou curativa. 18
 e docosahexaenóico")
19 Não são considerados fitofármacos compostos isolados que sofram qualquer etapa de semi-síntese ou modificação de sua estrutura química. XI - Ingestão Diária Recomendada (IDR): é a quantidade de proteína, vitaminas e minerais que deve ser consumida diariamente para atender às necessidades nutricionais da maior parte dos indivíduos e grupos de pessoas de uma população sadia, de acordo com o estabelecido na RDC nº 269, de 22 de setembro de 2005, da Anvisa, ou suas atualizações; XII - marcador: composto ou classe de compostos químicos (ex: alcalóides, flavonóides, ácidos graxos, etc.) presentes na matéria-prima vegetal, preferencialmente tendo correlação com o efeito terapêutico, que é utilizado como referência no controle da qualidade da matéria-prima vegetal e do medicamento fitoterápico; XIII - matéria-prima vegetal: compreende a planta medicinal, a droga vegetal ou o derivado vegetal; XIV - medicamento isento de prescrição médica: produto farmacêutico, tecnicamente obtido ou elaborado, com finalidade profilática, curativa ou paliativa, cujo grupo e as indicações terapêuticas estão descritos Resolução da Diretoria Colegiada nº 138, de 29 de maio de 2003, da Anvisa, que dispõe sobre o enquadramento na categoria de venda de medicamentos, ou suas atualizações, e ainda, para vitaminas e minerais, isolados ou associados entre si, com níveis de posologia diária abaixo do definido pela Portaria SVS/MS nº 40,de 16 de janeiro de 1998, que estabelece normas para níveis de dosagens diárias de vitaminas e minerais em medicamentos, ou suas atualizações; XV - nomenclatura botânica completa: espécie, autor do binômio, variedade, quando aplicável, e família; XVI - Nutrição Parenteral (NP): solução ou emulsão, composta basicamente de carboidratos, aminoácidos, lipídios, vitaminas e minerais, estéril e apirogênica, acondicionada em recipientes de vidro ou plástico, destinada à administração intravenosa em pacientes desnutridos ou não, em regime hospitalar, ambulatorial ou domiciliar, visando a síntese ou manutenção dos tecidos, órgãos ou sistemas; XVII - opoterápico: preparação obtida a partir de glândulas, tecidos, outros órgãos e secreções animais destinada a fim terapêutico ou medicinal; XVIII - perfil cromatográfico: padrão cromatográfico de constituintes característicos, obtido em condições definidas, que possibilite a identificação da espécie vegetal em estudo e a diferenciação de outras espécies; XIX - planta medicinal: espécie vegetal, cultivada ou não, utilizada com propósitos terapêuticos; XX - produtos para a prevenção da desidratação e para a manutenção da hidratação: soluções prontas para uso e/ou soluções concentradas para serem diluídas e/ou pós ou grânulos para diluição em água para prevenção da desidratação e para a manutenção da hidratação oral; XXI - própolis: produto de características físicas resinosas e composição variável, coletada a partir de várias espécies vegetais e que sofre adição de secreções da abelha, sendo classificada como opoterápico. 19

20 XXII - própolis específica: própolis com marcadores químicos definidos, diferenciados qualitativa e quantitativamente, conforme a região geográfica de origem. XXIII - proteínas: moléculas orgânicas constituídas por aminoácidos, atuam como catalisadores e contribuem com a sustentação estrutural da célula. XXIV - prospecção fitoquímica: testes de triagem, qualitativos ou semiquantitativos, que utilizam reagentes de detecção específicos para evidenciar a presença de grupos funcionais característicos na matéria-prima vegetal e que auxiliam na identificação da espécie vegetal e a diferenciação de outras espécies; XXV - sistema fechado: sistema de administração de solução parenteral que, durante todo o preparo e administração, não permite o contato da solução com o meio ambiente; XXVI - soluções para irrigação e soluções para diálise peritoneal: soluções em base aquosa, estéreis, apirogênicas, acondicionadas em recipiente único com capacidade de 100 ml ou mais, esterilizadas terminalmente; XXVII - Soluções Parenterais (SP): solução injetável, estéril e apirogênica, de grande ou pequeno volume, própria para administração por via parenteral; XXVIII - Soluções Parenterais de Grande Volume (SPGV): solução parenteral acondicionada em recipiente de dose única, em sistema fechado, com um volume nominal igual ou acima de 100 ml e até volume máximo de 1000 ml; XXIX - Soluções Parenterais de Pequeno Volume (SPPV): solução parenteral acondicionada em recipiente com a capacidade inferior a 100 ml; XXX - via parenteral: acesso para administração de medicamentos que alcancem espaços internos do organismo, incluindo vasos sanguíneos, órgãos e tecidos; CAPÍTULO II DA CATEGORIA DE MEDICAMENTOS ESPECÍFICOS Art. 5º Os seguintes produtos se enquadram para efeitos desta Resolução na categoria de medicamentos específicos: I - soluções para irrigação, diálise, enemas e expansores plasmáticos; II - Concentrados Polieletrolíticos para Hemodiálise (CPHD); III - nutrição parenteral; IV - soluções de grande e de pequeno volume, parenterais ou não, tais como, água para injeção, soluções de glicose, cloreto de sódio, demais compostos eletrolíticos ou açúcares e poliálcoois; V - opoterápicos isolados ou associados entre si e/ou a derivados vegetais e/ou vitaminas e/ou minerais e/ou aminoácidos e/ou proteínas e/ou fitofármaco; 20

21 VI - medicamentos à base de fitofármaco ou associações deste as vitaminas e/ou minerais e/ou aminoácidos e/ou proteínas; VII- medicamentos à base de rutina e/ou quercitina e/ou hesperidina e/ou diosmina e/ou troxerrutina e/ou cumarina, isolados ou associados entre si; VIII - produtos para a prevenção da desidratação e para a manutenção da hidratação; IX - antiácidos isolados ou associados entre si e/ou a antifiséticos, com exceção daqueles previstos na Lista de Medicamento Referência da Anvisa e na Notificação Simplificada de Medicamentos, conforme RDC nº 199, de 26 de outubro de 2006, que instituiu o Regulamento Técnico para a Notificação Simplificada de Medicamentos, ou suas atualizações; X - medicamentos à base de silimarina e/ou acetilmetionina e/ou metionina e/ou colina e/ou betaína e/ou ornitina e/ou acetil-cisteína e/ou ácidos biliares, isolados ou associados entre si, conforme finalidade de uso definida pelo Painel de Avaliação de Hepatoprotetores, publicado pela Anvisa, na RDC nº 41, de 26 de fevereiro de 2003, ou suas atualizações; XI - medicamentos à base de vitaminas e/ou minerais de uso tópico ou injetável; XII - medicamentos à base de vitaminas e/ou minerais e/ou aminoácidos e/ou proteínas isolados ou associados entre si, para uso oral, com pelo menos um dos componentes acima dos limites nutricionais estabelecidos pela IDR; XIII - medicamentos à base de derivados vegetais associados a vitaminas e/ou minerais e/ou aminoácidos e/ou proteínas e/ou fitofármaco; XIV - medicamentos de uso tópico à base de Cânfora, com exceção daqueles previstos na Notificação Simplificada de Medicamentos, conforme RDC nº 199, de 26 de outubro de 2006, que instituiu o Regulamento Técnico para a Notificação Simplificada de Medicamentos, ou suas atualizações; Parágrafo único. O medicamento que pertencer à categoria de medicamento específico não poderá solicitar registro junto à Anvisa como genérico, fitoterápico, dinamizado, similar, biológico ou novo. Art. 6º As vitaminas, minerais e aminoácidos, isoladas ou associadas, continuam também a ser regidas pela Portaria SVS/MS nº 40, de 16 de janeiro de 1998, que estabelece normas para níveis de dosagens diárias de vitaminas e minerais em medicamentos, ou suas atualizações; Parágrafo único. Caso a legislação brasileira não defina os valores dos Níveis Máximos (NM) de segurança dos produtos a que se refere o "caput" deste artigo, podem ser aplicados os níveis máximos de segurança de outros países, desde que seja apresentada a comprovação do nível máximo de segurança regulamentado através de códigos oficiais desses países. Art. 7º Os CPHD continuam também a ser regidos pela RDC nº 8, de 10 de janeiro de 2001, que aprovou o regulamento técnico que institui as Boas Práticas de Fabricação e Controle (BPFC) do Concentrado Polieletrolítico para Hemodiálise, ou suas atualizações 21
22 Art. 8º Os produtos para a prevenção da desidratação e para a manutenção da hidratação oral continuam também a ser regidos pela Portaria SVS/MS nº 108, de 25 de julho de 1991, que normatiza a composição de produtos para terapia de desidratação oral, de acordo com os conceitos de reidratação, manutenção e prevenção em terapia de reidratação oral contidos nas normas de controle de doenças diarréicas do Ministério da Saúde, ou suas atualizações. Art. 9º Os produtos para nutrição parenteral continuam também a ser regidos pela Portaria SVS/MS nº 272, de 08 de abril de 1998, que aprovou o regulamento técnico que fixa os requisitos mínimos exigidos para a terapia de NP, ou suas atualizações. Art. 10 Os medicamentos a base de silimarina e/ou acetilmetionina e/ou metionina e/ou colina e/ou betaína e/ou ornitina e/ou acetilcisteína e/ou ácidos biliares, isolados ou associados entre si, continuam também a ser regidos pelo anexo da RDC nº 41, de 26 de janeiro de 2003, da Anvisa, que publicou o Painel de avaliação de hepatoprotetores, ou suas atualizações. Seção I Das Medidas Antecedentes CAPÍTULO III DO REGISTRO DE PRODUTOS NACIONAIS Art. 11 Previamente à apresentação do processo de registro de medicamento específico, a empresa interessada deverá notificar a produção de lotes-piloto, de acordo com o Guia para a notificação de lotes piloto de medicamentos, publicado pela Anvisa na Instrução Normativa - IN nº 02, de 30 de março de 2009, ou suas atualizações. Parágrafo único. O disposto no "caput" deste artigo não se aplica aos produtos importados. Seção II Da Documentação Art. 12 Todos os documentos para registro deverão ser encaminhados na forma de uma via impressa numerada e rubricada em todas as folhas pelo responsável técnico pela empresa. Parágrafo único. Acompanhando a documentação, deverá ser apresentada a folha de rosto, conforme modelo disposto no Anexo II desta Resolução, e índice com numeração das respectivas páginas das documentações. Adicionar ao processo cópia de especificações, métodos analíticos, referências bibliográficas e, quando aplicável, va- lidação de metodologia analítica em mídia eletrônica, com arquivos em formato aceito pela Anvisa. Art. 13 Toda a documentação deverá ser apresentada em idioma português, acompanhada da documentação original, quando não se tratar de tradução juramentada na forma da lei. 22
23 Art. 14 A empresa deverá protocolar um processo para cada medicamento específico, com relatórios separados para cada forma farmacêutica, apresentando os seguintes documentos: I - formulários de petição (FP); II - via original do comprovante de recolhimento da taxa de fiscalização de vigilância sanitária, ou isenção, quando for o caso; III - cópia da licença de funcionamento da empresa (alvará sanitário), atualizada, ou protocolo da solicitação da renovação da referida licença; IV - cópia do CRT, atualizado, emitido pelo Conselho Regional de Farmácia; V - cópia do protocolo da notificação da produção de lotes piloto; VI - cópia do CBPFC, atualizado, emitido pela Anvisa para a linha de produção na qual o medicamento especifico será fabricado; e VII - relatório técnico. Seção III Relatório Técnico Art. 15 O relatório técnico deve conter as seguintes in- formações: I - relatório de estabilidade do medicamento; II - dados do derivado vegetal, do fitofármaco, do derivado de síntese ou semi-síntese e do opoterápico, quando presentes; III - layout das embalagens primária e secundária, modelo de bula, e rótulo, conforme legislação vigente; IV - documentação referente a cada local de fabricação, caso a empresa solicite o registro em mais de um local de fabricação; V - relatório de produção; VI - controle de qualidade; e VII - relatório técnico com informações de segurança e eficácia, quando aplicável. Seção IV 23
24 Relatório de Estabilidade Art. 16 A empresa deverá apresentar resultados do estudo de estabilidade acelerado concluído acompanhado do estudo de estabilidade de longa duração em andamento de 3 (três) lotes-piloto, ou estudos de estabilidade de longa duração já concluídos, todos de acordo com a Resolução - RE nº 01, de 29 de julho de 2005, da Anvisa, que publicou o Guia para a realização de estudos de estabilidade de medicamentos, ou suas atualizações. 1º Decorrido o prazo de validade declarado para o medicamento, a empresa deverá protocolar, na forma de complementação de informações ao processo, relatório de resultados e avaliação final do estudo de estabilidade de longa duração dos três lotes apresentados no pedido de registro, de acordo com o cronograma previamente apresentado, assim como a declaração do prazo de validade e cuidados de conservação definitivos, sob pena de configuração de infração sanitária. 2º Para medicamentos com três ou mais concentrações e formulações proporcionais, a empresa deverá apresentar os resultados do estudo de estabilidade das concentrações menor e maior. 3º Para medicamentos acondicionados em embalagens de volumes diferentes serão aceitos os estudos de estabilidade do maior e menor volume, desde que comprovem as mesmas características, de acordo com o Guia de estabilidade reduzido publicado no sítio eletrônico da Agência Nacional de Vigilância Sanitária. Seção V Relatório de Produção e Controle de Qualidade Art. 17 O relatório de produção deve conter as seguintes informações: I - forma farmacêutica; II - descrição detalhada da fórmula conforme a Denominação Comum Brasileira (DCB) ou, em sua ausência, a Denominação Comum Internacional (DCI) ou a denominação utilizada no Chemical Abstracts Service (CAS); III - descrição da quantidade de cada componente expresso no Sistema Internacional de unidades (SI) por unidade farmacotécnica, indicando sua função na fórmula; IV - tamanhos mínimo e máximo dos lotes industriais a serem produzidos; V - descrição de todas as etapas do processo de produção, contemplando os equipamentos utilizados; VI - metodologia de controle do processo produtivo; e VII - descrição dos critérios de identificação do lote industrial Art. 18 O relatório de controle de qualidade deve apresentar as seguintes informações gerais: I - controle da Encefalopatia Espongiforme Transmissível (EET) de acordo com a legislação vigente; 24
25 II - laudo de análise de todas as matérias-primas utilizadas e do produto final, contendo as especificações empregadas; III - referências farmacopeicas consultadas e reconhecidas pela Anvisa, de acordo com a legislação vigente; e IV - especificação do material de embalagem primária do medicamento. 1º Quando não forem utilizadas referências farmacopeicas reconhecidas pela Anvisa, deve ser apresentada descrição detalhada de todas as metodologias utilizadas no controle de qualidade, com métodos analíticos validados de acordo com a Resolução - RE nº 899, de 29 de maio de 2003, da Anvisa, que publicou o "Guia de validação de métodos analíticos e bioanalíticos", ou suas atualizações, indicando a fonte de desenvolvimento. 2º Em caso de SPGV, a empresa deve enviar laudo de análise da embalagem primária, conforme ensaios preconizados na RDC nº 29, de 17 de abril de 2007, da Anvisa, que dispõe sobre as regras referentes ao registro e comercialização para a substituição do sistema de infusão aberto para fechado em SPGV, ou suas atualizações. Art. 19 Os testes referentes ao controle da qualidade do medicamento específico, quando terceirizados, devem atender ao preconizado na RDC nº 25, de 29 de março de 2007, da Anvisa, que dispõe sobre as regras referentes à terceirização de etapas de produção, análises de controle de qualidade e de armazenamento de medicamentos, ou suas atualizações. Subseção I Do Derivado Vegetal Art. 20 Quando a empresa fabricante do medicamento específico for também produtora do derivado vegetal, deve ser apresentado laudo de análise da droga vegetal, indicando o método utilizado, especificação e resultados obtidos para um lote dos ensaios abaixo descritos. I - testes de autenticidade, caracterização organoléptica, identificação macroscópica e microscópica; II - descrição da droga vegetal em farmacopéias reconhecidas pela Anvisa, ou, em sua ausência, publicação técnicocientífica indexada ou laudo de identificação emitido por profissional habilitado III - testes de pureza e integridade, incluindo: a) cinzas totais e/ou cinzas insolúveis em ácido clorídrico; b) umidade e/ou perda por dessecação; c) pesquisa de matérias estranhas; d) pesquisa de contaminantes microbiológicos; e e) pesquisa de metais pesados; 25
26 IV - método de estabilização, quando empregado, secagem e conservação utilizada, com seus devidos controles, quando cabível; V - método para eliminação de contaminantes, quando empregado, e a pesquisa de eventuais alterações; VI - avaliação da ausência de aflatoxinas, a ser realizada quando citada em monografia específica em farmacopéia reconhecida ou quando existir citação em literatura científica da necessidade dessa avaliação ou de contaminação da espécie por aflatoxinas; VII - local de coleta; VIII - perfil cromatográfico ou prospecção fitoquímica; e IX - análise quantitativa do(s) marcador(es) ou controle biológico. Art. 21 O relatório de controle de qualidade deve apresentar laudo de análise do derivado vegetal, indicando o método utilizado, especificação e resultados obtidos para um lote dos ensaios abaixo descritos: I - solventes, excipientes e/ou veículos utilizados na extração do derivado; II - relação aproximada droga vegetal: derivado vegetal; III - testes de pureza e integridade, incluindo: a) pesquisa de contaminantes microbiológicos; b) pesquisa de metais pesados; e c) resíduos de solventes (para extratos que não sejam obtidos por etanol e/ou água); IV - método para eliminação de contaminantes, quando empregado, e a pesquisa de eventuais alterações; V - caracterização físico-química do derivado vegetal incluindo: a) caracterização organoléptica, resíduo seco, ph, teor alcoólico e densidade (para extratos líquidos); b) umidade/perda por dessecação, solubilidade e densidade aparente (para extratos secos); c) densidade, índice de refração, rotação óptica (para óleos essenciais); e d) índice de acidez, de éster, de iodo (para óleos fixos); VI - avaliação da ausência de aflatoxinas, a ser realizado quando citado em monografia específica em Farmacopéia reconhecida ou quando existir citação em literatura científica da necessidade dessa avaliação ou de contaminação da espécie por aflatoxinas; VII - perfil cromatográfico ou prospecção fitoquímica; e 26
27 VIII - análise quantitativa do(s) marcador(es) ou controle biológico. Parágrafo único. Outros testes podem ser adicionados ou substituir os descritos no inciso V de acordo com monografia farmacopeica respectiva. Art. 22 Quando a empresa não for a produtora do derivado vegetal, deverá enviar laudo de análise do fornecedor, contendo as seguintes informações: I - nomenclatura botânica completa; II - parte da planta utilizada; III - solventes, excipientes e/ou veículos utilizados na extração do derivado; IV - relação aproximada droga vegetal:derivado vegetal; e V - descrição do método para eliminação de contaminantes, quando utilizado, e a pesquisa de eventuais alterações. Art. 23 O relatório de controle de qualidade deve apresentar laudo de análise do produto acabado indicando o método utilizado, especificação e resultados obtidos para um lote, dos ensaios abaixo descritos: I - perfil cromatográfico ou prospecção fitoquímica; II - análise quantitativa do(s) marcador(es) específico(s) de cada espécie ou controle biológico; III - resultados de todos os testes realizados no controle da qualidade para um lote do medicamento de acordo com a forma farmacêutica solicitada; IV - especificações do material de embalagem primária; e V - controle dos excipientes utilizados na fabricação do medicamento por método estabelecido em farmacopéia reconhecida. 1º Para associações de espécies vegetais em que a determinação quantitativa de um marcador por espécie não é possível, poderá(ão) ser apresentado(s) o(s) perfil(is) cromatográfico(s), que contemple(m) a presença de ao menos um marcador específico para cada espécie na associação, complementado pela determinação quantitativa do maior número possível de marcadores específicos para cada espécie. 2º A impossibilidade técnica de determinação quantitativa de um marcador para cada espécie da associação deve ser devidamente justificada. 3º Na hipótese do ensaio descrito no inciso V, não sendo uma farmacopéia reconhecida pela Anvisa, a empresa deve descrever detalhadamente todas as metodologias utilizadas no controle de qualidade. Subseção II Do Fitofármaco, Derivado de Síntese ou Semi-síntese 27
28 Art. 24 Quando a empresa fabricante do medicamento específico for também a produtora do fitofármaco, deverá ser apresentado relatório descritivo que contemple as etapas de extração, isolamento e purificação do fitofármaco, metodologia, equipamentos, solventes e/ou excipientes utilizados. 1º Deverá ser enviado laudo de análise do fitofármaco que contemple os requisitos mínimos de identidade e qualidade, conforme monografia farmacopeica reconhecida pela Anvisa. 2º Na ausência de monografia farmacopeica reconhecida pela Anvisa, deverão ser apresentadas as referências bibliográficas das fontes de desenvolvimento da metodologia analítica, junto às informações do fabricante do insumo ativo que identifique os requisitos de qualidade adotados. Art. 25 Quando a empresa fabricante do medicamento específico não for a produtora do fitofármaco, deverá ser enviado laudo do fornecedor contendo a descrição dos solventes, excipientes e/ou veículos utilizados para obtenção do fitofármaco. Art. 26 A empresa solicitante de registro de medicamento específico, cuja substância ativa esteja identificada no inciso VII do Art. 5º desta Resolução e seja derivada de síntese ou semi-síntese, deverá apresentar cópia da documentação, abaixo discriminada, em papel timbrado da empresa fabricante do fármaco. I - relatório descritivo contendo dados gerais da empresa fabricante do fármaco, inclusive o endereço completo da mesma, além das etapas de síntese envolvidas, metodologia, equipamentos, solventes, excipientes e/ou reagentes utilizados; II - rota de síntese do fármaco, com descrição das moléculas intermediárias, seus nomes químicos e solventes utilizados e com determinação dos pontos críticos da produção e ensaios de controle em processos bem definidos; III - laudo de análise do fármaco, com descrição das metodologias e referências empregadas no controle de qualidade, de acordo com os requisitos mínimos de identidade e qualidade adotados; IV - quantificação dos contaminantes, de acordo com a rota de síntese do fármaco; V - metodologia analítica adotada e resultados dos testes de determinação dos prováveis polimorfos do fármaco, no caso de fármacos que apresentem polimorfismo; e VI - dados sobre os teores dos estereoisômeros, cuja proporção possa comprometer a eficácia e a segurança do medicamento, no caso de fármacos que apresentam quiralidade; Parágrafo único. Fica facultado ao fabricante do fármaco enviar diretamente a Anvisa a documentação explicitada neste artigo, devidamente identificada com o número do processo a que se relaciona. Subseção III Do Opoterápico Art. 27 Quando a empresa fabricante do medicamento específico for também a empresa produtora do opoterápico, deverá apresentar relatório descritivo contendo etapas de produção da preparação de origem animal, metodologia, equipamentos, solventes e/ou 28
29 excipientes utilizados. Parágrafo único. Deverá ser apresentado laudo de análise que contemple os requisitos mínimos de identidade e qualidade validados da preparação farmacêutica de origem animal. Art. 28 Quando a empresa fabricante do medicamento específico não for a empresa produtora do opoterápico, deve ser apresentado laudo de análise do fornecedor que contemple os requisitos mínimos de identidade e qualidade validados. 1º Para os ácidos graxos ômega-3, deverá ser apresentado laudo de análise, conforme requisitos mínimos de identidade e qualidade definidos pela Farmacopéia Européia, em sua última edição, ou outro compêndio oficial reconhecido pela Anvisa, de acordo a real tipificação do ácido graxo ômega-3 utilizado. 2º No laudo de análise descrito no 1º, deverá ser indicado a referência do método empregado, a especificação e resultados obtidos para um lote dos ensaios abaixo descritos: I - características físico-químicas incluindo: a) características organolépticas; b) solubilidade; c) absorbância; e d) material insaponificável. II - testes de pureza e integridade incluindo: a) pesquisa de anisidina, peróxido, oligômeros, estearina, resíduos de solvente e resíduos de pesticida; b) pesquisa de metais pesados: mercúrio, cádmio, chumbo e arsênio; c) pesquisa de contaminantes microbiológicos; d) pesquisa de dioxinas, furanos e bifenilpoliclorados. III - identificação; e IV - doseamento. Art. 29 Quando a empresa fabricante do medicamento específico for também a produtora do extrato de própolis, deverá ser apresentado laudo de análise que contemple os requisitos mínimos de identidade e qualidade validados, contendo as seguintes informações: a) Própolis in natura 29
30 I - características sensoriais: aspecto, cor, sabor e odor; II - requisitos físico-químicos: perda por dessecação, teor de cinzas totais, cinzas insolúveis em ácido clorídrico; III - determinação de: solúveis em etanol, teor de ceras, teor de fenóis totais, teor de flavonóides, análise qualitativa de marcadores específicos (perfil cromatográfico ou prospecção fitoquímica), análise quantitativa de marcadores específicos ou controle biológico; IV - contaminantes: pesquisa e identificação de patógenos, coliformes, fungos e leveduras, metais pesados, determinação de material estranho; V - informações sobre a espécie da abelha e as espécies da flora presentes no local da colméia onde foi coletada a própolis. b) Extrato de própolis I - características organolépticas: aspecto, cor, sabor e odor; II - requisitos físico-químicos: a) extrato líquido: determinação do extrato seco, densidade, teor alcoólico e ph; b) extrato seco: umidade, perda por dessecação e densidade aparente; III - determinação de: teor de fenóis totais, teor de flavonóides, análise qualitativa de marcadores específicos (perfil cromatográfico ou prospecção fitoquímica), análise quantitativa de marcadores específicos ou controle biológico; IV - contaminantes: pesquisa e identificação de patógenos, coliformes, fungos e leveduras, metais pesados, determinação de material estranho; Art. 30 Quando a empresa fabricante do medicamento específico não for a produtora do extrato de própolis, deverá ser apresentado laudo de análise do fornecedor, contendo as informações descritas acima para o extrato acompanhado da descrição da espécie da abelha e das espécies da flora específica presentes no local da colméia e compatíveis com o raio de atuação da abelha. Seção VI Dos Modelos de Bula, Rótulo e Embalagem Art. 31 A empresa deverá apresentar modelo de bula e layout das embalagens primária e secundária do medicamento, conforme legislação específica. 1º Para as associações contendo derivados vegetais, opoterápicos, vitaminas, minerais, aminoácidos, proteínas, fitofármacos estão mantidas as obrigações de tabela informativa do quantitativo percentual da IDR, quando estabelecido, conforme legislação específica. 30
31 2º Para os medicamentos que contenham ácidos graxos ômega-3, deverão ser informados, junto à tabela informativa do quantitativo da IDR dos nutrientes, os valores de gordura poli-insaturada ômega-3 total, além do quantitativo dos ácidos eicosapentaenóico - EPA e docosahexaenóico - DHA. Seção VII Da Segurança e Eficácia Art. 32 O relatório técnico deve conter informações sobre segurança e eficácia comprovadas por: I - relatório de segurança e eficácia pré-clínica e clínica; ou II - dados de literatura técnico-científica que contemple essas informações; ou III - tradicionalidade de uso; Art. 33 Estão isentos da comprovação de eficácia e segurança: I - medicamentos à base de associações entre vitaminas e/ou minerais e o derivado vegetal de Panax ginseng C. A. Mey, com padronização de marcador e posologia diária definida pela Lista de Medicamentos de Registro Simplificado, publicada pela Instrução Normativa - IN nº 05, de 11 de dezembro de 2008, da Anvisa, ou suas atualizações, cujos níveis de dosagem diária para vitaminas e minerais estejam abaixo do definido pela Portaria SVS/MS nº 40, de 16 de janeiro de 1998, ou suas atualizações, com indicação terapêutica para a prevenção e recuperação em casos de fadiga física e psíquica, atuando como adaptógeno, e suplementação vitamínico-mineral; II - medicamentos à base de própolis de uso tópico, na cavidade bucal, com as indicações de uso: como antiinflamatório, anti-séptico e cicatrizante; e III - medicamentos à base de vitaminas e/ou minerais e/ou aminoácidos, isolados ou associados entre si, de uso oral classificados como medicamentos isentos de prescrição médica; V - produtos para a prevenção da desidratação e para a manutenção da hidratação; VI - Os CPHD conforme regido pela Resolução da Diretoria Colegiada - RDC nº 8, de 10 de janeiro de 2001, que aprovou o regulamento técnico que institui as BPFC do CPHD, ou suas atualizações. Art. 34 Para os medicamentos à base de vitaminas e/ou minerais de uso tópico classificados como medicamentos isentos de prescrição médica, além de outros à base de cânfora, deverão ser apresentados dados de literatura técnicocientífica que suportam afinalidade terapêutica pretendida para a associação. Art. 35 Para os antiácidos isolados ou associados a antifiséticos será considerada a quantidade de cada íon e sua capacidade neutralizante, devendo ser apresentados dados de literatura técnico-científica que suportam as doses pretendidas, junto à justificativa 31
32 técnico-científica de racionalidade da associação. Art. 36 Para os medicamentos à base dos hepatoprotetores identificados no inciso X do Art. 5º desta Resolução devem ser apresentados dados de literatura técnico-científica indexada dos componentes ativos isolados nas dosagens pretendidas. Art. 37 Para os medicamentos à base de vitaminas e/ou minerais sob prescrição médica, para os opoterápicos isolados ou associados a vitaminas e/ou minerais e/ou aminoácidos deve ser apresentado estudo de eficácia e segurança clínica ou dados da literatura que comprovem a eficácia e segurança da associação, nas doses pretendidas, através de estudos clínicos publicados em literatura técnico-científica indexada e justificativa técnico-científica de racionalidade da associação. 1º Para os medicamentos à base de ácidos graxos ômega-3 associados a vitaminas e/ou minerais, para uso oral, com níveis de dosagem diária abaixo do definido pela Portaria SVS/MS nº 40, de 16 de janeiro de 1998, ou suas atualizações, sem prescrição médica, de acordo com o estabelecido pela RDC nº 138, de 29 de maio de 2003, ou suas atualizações, não serão exigidos estudos de comprovação de eficácia clínica. 2º Para os medicamentos à base de própolis com as indicações terapêuticas ou forma de uso diferente daquelas descritas no inciso II do Art. 33 desta Resolução, deverá ser apresentado estudo de eficácia e segurança clínica de uso do medicamento ou dados da literatura que comprovem a eficácia e segurança, através de estudos clínicos publicados em literatura técnicocientífica indexada, considerando a própolis específica utilizada 3º Os estudos com a própolis específica somente serão válidos para produtos baseados nela própria. 4º Para os medicamentos à base de opoterápicos isolados ou associados entre si e/ou a derivados vegetais e/ou vitaminas e/ou minerais e/ou aminoácidos e/ou proteínas e/ou fitofármacos os requisitos para comprovação de segurança e eficácia encontram-se estabelecidos na Tabela I do Anexo I desta Resolução. Art. 38 Para os medicamentos à base de associações definidas pelo inciso XIII e do art. 5º desta Resolução, bem como aqueles à base dos ativos: rutina e/ou quercitina e/ou hesperidina e/ou diosmina, além de outros à base da associação ativa troxerrutina e cumarina, os requisitos para comprovação de segurança e eficácia encontram-se estabelecidos na Tabela I do Anexo I desta Resolução. Parágrafo Único. Para finalidade terapêutica diferente daquelas apresentadas na Tabela I do Anexo I, a empresa deverá enviar o relatório de segurança e eficácia clínica para o medicamento e justificativa técnico-científica de racionalidade da associação. Art. 39 Para comprovação de segurança e eficácia pela tradicionalidade de uso do medicamento específico deverão ser apresentadas publicações técnico-científicas que serão avaliadas conforme os seguintes critérios: I - indicação de uso episódico ou para curtos períodos de tempo; 32
33 II - indicação para doenças de baixa gravidade ou relacionada à melhoria ou manutenção da saúde; III - coerência das indicações terapêuticas propostas com as comprovadas pelo uso tradicional; IV - ausência de grupos ou substâncias químicas tóxicas, ou presentes dentro de limites comprovadamente seguros; V - comprovação de continuidade de uso seguro por período igual ou superior a 10 anos no Brasil; e VI - racionalidade das associações de ativos. 1º Não serão permitidas alterações das seguintes características do medicamento durante o período de comercialização igual ou superior a 10 anos: substância(s) ativa(s): qualitativa e quantitativa, forma farmacêutica incluindo sistema de liberação, posologia e indicação terapêutica. 2º Deverá ser apresentado o Documento de Descrição do Sistema de Farmacovigilância (DDSF) e Relatório Periódico de Farmacovigilância (RPF) para o medicamento, de acordo com a regulamentação sanitária em vigor. 3º Deverão ser apresentados material de bula, embalagem e de fins publicitários do medicamento que comprove que o produto fora utilizado durante o período mínimo de comercialização definido no inciso V do "caput" deste artigo, para a indicação terapêutica proposta. Parágrafo único. Para os medicamentos específicos que comprovarem segurança e eficácia por tradicionalidade de uso, deve ser inserida a seguinte frase na bula, embalagem e material publicitário: "Medicamento registrado com base no uso tradicional, não sendo recomendado seu uso por período prolongado". CAPÍTULO IV Do Registro de Produtos Importados Art. 40 Os fabricantes ou seus representantes que pretenderem comercializar medicamentos específicos produzidos em território estrangeiro, além de cumprir os requisitos dessa Resolução referentes à fabricação nacional, devem apresentar: I - autorização da empresa fabricante, detentora do registro e/ou da marca, para o registro, representação comercial ou uso da marca no Brasil, quando aplicável; II - cópia do CBPFC emitido pela Anvisa para a empresa fabricante, atualizado, por linha de produção; III - cópia do CBPFC emitido pela Anvisa ou do protocolo do pedido de inspeção para este fim, para a linha de produção da empresa requerente do registro, quando se tratar de importação de produto a granel ou em sua embalagem primária; IV - laudo de análise com especificação e referência bibliográfica, ou descrição de metodologia de controle da qualidade físico-química, química, microbiológica e biológica que o importador realizará, de acordo com a forma farmacêutica e apresentação: produto acabado, a granel ou na embalagem primária; 33
34 V - comprovação do registro do produto, emitida pelo órgão responsável pela vigilância sanitária do país de origem. 1º Na impossibilidade do cumprimento do disposto no inciso V deste artigo, deverá ser apresentada comprovação de registro em vigor, emitida pela autoridade sanitária do país em que seja comercializado ou autoridade sanitária internacional e aprovado em ato próprio da Anvisa. 2º No caso de a Anvisa ainda não ter realizado inspeção na empresa fabricante, será aceito comprovante do pedido de inspeção sanitária à Anvisa, acompanhado de cópia do CBPFC de produtos farmacêuticos por linha de produção, emitido pelo órgão responsável pela vigilância sanitária do país fabricante. 3º A Anvisa poderá, conforme legislação específica, efetuar a inspeção da empresa fabricante no país ou bloco de origem. Art. 41 Deve ser enviada à Anvisa cópia dos resultados e da avaliação do teste de estabilidade na embalagem primária de comercialização, de acordo com a Resolução - RE nº 01, de 29 de julho de 2005, da Anvisa, que publicou o Guia para a Realização de Estudos de Estabilidade de Medicamentos. Art. 42 O prazo de validade do produto importado a granel deve ser contado a partir da data de fabricação do produto no exterior, e não da data de embalagem no Brasil, respeitando o prazo de validade registrado na Anvisa. Art. 43 Todo o material relativo ao produto, tais como os relatórios de produção e controle da qualidade, e as informações contidas em rótulos, bulas e embalagens deve estar em idioma português, atendendo à legislação em vigor. Art. 44 Os documentos oficiais em idioma estrangeiro, usados para fins de registro, expedidos pelas autoridades sanitárias, devem ser acompanhados de tradução juramentada na forma da lei. Art. 45 Havendo necessidade de importar amostras, a empresa deve solicitar à Anvisa a devida autorização para a importação. Art. 46 Decorrido o prazo de validade declarado para o medicamento, a empresa deverá protocolar, na forma de complementação de informações ao processo, relatório de resultados e avaliação final do estudo de estabilidade de longa duração dos 3 (três) lotes apresentados no pedido de registro, de acordo com o cronograma previamente apresentado, assim como a declaração do prazo de validade e cuidados de conservação definitivos, sob pena de configuração de infração sanitária. Seção I CAPÍTULO IV DAS MEDIDAS PÓS-REGISTRO Das Alterações, Inclusões, Suspensão, Reativação e Cancelamento Pós-Registro 34
35 Art. 47 As alterações, inclusões, suspensão, reativação e cancelamento pós-registro de medicamento específico devem seguir os procedimentos especificados na RDC nº 48, de 6 de outubro de 2009, da Anvisa, que dispõe sobre a realização de "alterações, inclusões, suspensão, reativação e cancelamento pós-registro de medicamentos", ou suas atualizações. Parágrafo único. Para as alterações pós-registro de fabricante de fármaco, de acordo com o estabelecido no Art. 26 desta Resolução, deverão ser obedecidos os procedimentos especificados para o assunto, na RDC nº 48, de 6 de outubro de 2009, ou suas atualizações. Seção II Da Renovação de Registro Art. 48 Todas as empresas, no primeiro semestre do último ano do qüinqüênio de validade do registro, deverão apresentar à Anvisa os seguintes documentos para efeito de renovação: I - Formulário de Petição - FP devidamente preenchido; II - via original do comprovante de recolhimento da taxa de fiscalização de vigilância sanitária ou de isenção, quando for o caso; III - cópia do CRT, atualizado, emitido pelo Conselho Regional de Farmácia; IV - cópia da licença de funcionamento da empresa (alvará sanitário), atualizada, ou protocolo da solicitação da renovação da referida licença; V - cópia do CBPFC, atualizado, para a linha de produção na qual o produto classificado como medicamento específico será fabricado; VI - demonstração da existência de um sistema de farmacovigilância na empresa para monitoração de falhas terapêuticas e efeitos colaterais indesejáveis, de acordo com legislação específica; VII - última versão de layout de bula, rótulo e embalagem que acompanha o produto; VIII - lista com as alterações do produto que contemple todas as alterações e/ou inclusões pós-registro ocorridas durante o último período de validade do registro do produto, acompanhados de cópia do DOU, ou na ausência, cópia do(s) protocolo(s) da(s) petição(ões) correspondente(s); IX - relatórios de segurança e eficácia e relatórios de produção, controle da qualidade, conforme determinado por essa Resolução, caso não tenham sido previamente apresentados; X - resultados do estudo de estabilidade de acompanhamento, de acordo com o Guia de Estabilidade de Medicamentos, publicado pela Anvisa na RE nº 01, de 29 de julho de 2005, ou suas atualizações; e XI - cópia de notas fiscais comprovando a comercialização do medicamento em, no mínimo, uma nota por forma farmacêutica. 35
36 1º Poderá ser apresentada uma declaração referente às apresentações comerciais não comercializadas para as quais a empresa tenha interesse em manter o registro, desde que pelo menos uma apresentação daquela forma farmacêutica tenha sido comercializada. 2º Devem ser enviados relatórios periódicos de farmacovigilância, de acordo com a legislação específica. Art. 49 Para a renovação do registro de produtos importados deverão ser apresentados, além do disposto no art. 48 desta Resolução, laudo de análise de três lotes importados nos últimos três anos do controle da qualidade físicoquímica, química, microbiológica e biológica, de acordo com a forma farmacêutica, realizados pelo importador no Brasil. CAPÍTULO V Das Disposições Finais e Transitórias Art. 50 Para os medicamentos registrados em outras categorias, a adequação a esta resolução deverá ocorrer no momento da renovação de registro do produto. 1º Para as petições que estejam protocoladas na Anvisa, serão concedidas três meses para protocolo de adequações necessárias ao cumprimento do disposto nesta Resolução, contados a partir da data de sua publicação. 2º Será aceita a adequação de formulações com supressão de ativos, desde que comprovada segurança, eficácia e qualidade para a nova formulação, nos termos desta Resolução. 3º Serão concedidos 12 meses de prazo para protocolo das adequações que tratam do relatório de estabilidade para as novas formulações, a partir da data de publicação desta Resolução. 4º Para os casos em que as alterações da formulação impliquem em novos estudos de segurança e eficácia para o medicamento, serão concedidos 18 meses de prazo para protocolo do relatório conclusivo, nos termos dispostos nesta Resolução, a partir da data de sua publicação. Art. 51 A Anvisa poderá realizar análise de controle de lotes comercializados para monitoração da qualidade e da conformidade do medicamento com as informações apresentadas no registro ou renovação de registro. Art. 52 A Anvisa poderá, a qualquer momento, exigir provas adicionais relativas à identidade e qualidade dos componentes, da segurança e da eficácia de um medicamento, em caso de dúvidas ou ocorrências que deem ensejo a avaliações complementares, mesmo após a concessão do registro. Art. 53 O descumprimento das disposições contidas nesta Resolução e no regulamento por ela aprovado constitui infração sanitária, nos termos da Lei nº 6.437, de 20 de agosto de 1977, sem prejuízo das responsabilidades civil, administrativa e penal cabíveis. Art. 54 Fica revogada a Resolução de Diretoria Colegiada da Anvisa - RDC nº 132, de 29 de maio de Art. 55 Esta Resolução entra em vigor na data da sua publicação. 36
37 RDC Nº 48, DE 6 DE OUTUBRO DE 2009 Dispõe sobre realização de alteração, inclusão, suspensão, reativação, e cancelamento pós-registro de medicamentos e dá outras providências. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto Nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria Nº 354 da Anvisa, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada em 5 de outubro de 2009, considerando as diretrizes, as prioridades e as responsabilidades estabelecidas na Política Nacional de Medicamentos, instituída pela Portaria n 3.916/MS/GM, de 30 de outubro de 1998, que busca garantir condições para segurança e qualidade dos medicamentos consumidos no país e promover o uso racional e o acesso da população àqueles considerados essenciais; considerando a competência da Anvisa para regulamentar os produtos e serviços que envolvam risco à saúde pública, estabelecida na Lei Nº 9.782, de 26 de janeiro de 1999, e especialmente no inciso I do 1º de seu art. 8º, que inclui os medicamentos de uso humano, suas substâncias ativas e demais insumos, processos e tecnologias entre os bens e produtos submetidos ao controle e fiscalização sanitária pela Agência; considerando que o registro dos produtos de que trata a Lei n.º 6.360, de 23 de setembro de 1976, poderá ser objeto de regulamentação pela Anvisa, visando a desburocratização e a agilidade nos procedimentos, nos termos do art. 41 da Lei n.º 9.782, de 1999, desde que isto não implique riscos à saúde da população ou à condição de fiscalização das atividades de produção e circulação; considerando que a atividade da Anvisa deve ser juridicamente condicionada pelos princípios da legalidade, celeridade, finalidade, razoabilidade, impessoalidade, imparcialidade, publicidade, moralidade e economia processual, nos termos do art. 29 do Regulamento da Anvisa aprovado pelo Decreto n.º 3.029, de 1999; considerando as disposições contidas na Lei n.º 6.360, de 1976, e no Decreto n.º , de 5 de janeiro de 1977, acerca do sistema de vigilância sanitária a que ficam sujeitos os medicamentos; adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Fica aprovado o Regulamento Técnico que estabelece os requisitos mínimos para os procedimentos de alteração, inclusão, suspensão, reativação e cancelamento pós-registro de medicamentos, nos termos desta Resolução. Objetivo Capítulo I Seção I Art. 2º Este Regulamento tem o objetivo de classificar as modificações pós-registro de medicamentos e estabelecer a documentação e os ensaios exigidos pela Anvisa. 37
38 Seção II Abrangência Art. 3º Este Regulamento aplica-se a medicamentos específicos, genéricos, novos e similares já registrados. Definições Seção III Art. 4º Para efeito deste Regulamento Técnico, são adotadas as seguintes definições: I. Histórico de Mudanças do Produto (HMP): Formulário no qual deverão ser registradas as mudanças/alterações ou inclusões pósregistro de medicamentos. Algumas mudanças consideradas de menor impacto, conforme definidas nesta norma, serão registradas somente neste histórico e isentas de protocolização individual. II. Protocolo de estudo de estabilidade: Documento por meio do qual se define o plano de estudo de estabilidade incluindo as provas e critérios de aceitação, cronograma, características do lote a ser submetido ao estudo, quantidade das amostras, condições do estudo, métodos analíticos e material de acondicionamento. III. Mudanças múltiplas concomitantes: São mudanças decorrentes de uma solicitação principal de acordo com o escopo deste regulamento. Quando permitidas nesta norma, poderão ser realizadas concomitantemente à mudança principal sem necessidade de protocolização adicional. IV. Mudanças múltiplas paralelas: Protocolização conjunta de duas ou mais solicitações de mudanças diretamente relacionadas e que ocorrem simultaneamente. DAS DISPOSIÇÕES INICIAIS Capítulo II Art. 5º A Anvisa concede, para os seguintes assuntos desta Resolução, autorização prévia para a implementação imediata, mediante protocolo de petição ou anotação no Histórico de Mudanças do Produto, conforme disposto nos termos dos capítulos específicos deste Regulamento. I. Alteração ou inclusão de local de embalagem secundária; II. Alteração ou inclusão de local de embalagem primária; III. Alteração menor do processo de produção; IV. Alteração ou inclusão de equipamento de embalagem primária; V. Alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento; VI. Inclusão de tamanho de lote em até 10 vezes; VII. Alteração menor de excipiente; 38
39 VIII. Adequação de especificações e métodos analíticos a compêndio oficial ou estreitamento de faixa de especificação; IX. Exclusão de local de fabricação do fármaco ou local de embalagem primária ou local de embalagem secundária ou local de fabricação do produto; X. Redução do prazo de validade com manutenção dos cuidados de conservação. 1º A implementação imediata das adequações, alterações, exclusões, inclusões ou reduções relacionadas neste artigo não impede a análise, a qualquer tempo, da documentação exigida, quando as alterações solicitadas poderão ser deferidas ou indeferidas. 2º As alterações não relacionadas neste artigo só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outras regras específicas para cada petição. Art. 6º Toda a documentação deve estar de acordo com legislação específica e, existindo guia específico este deverá ser atendido integralmente. Art. 7º Todas as petições de alterações, inclusões, suspensões, reativações, e cancelamentos pós-registro que necessitem de protocolização deverão estar acompanhadas dos seguintes documentos: caso; I. Via original de recolhimento de taxa de fiscalização de vigilância sanitária ou de isenção, quando for o II. Formulários de Petição - FP1 e FP2, devidamente preenchidos; I. III. Justificativa da solicitação contemplando a descrição detalhada e o racional da proposta, conforme Anexo Art. 8º O Histórico de Mudanças do Produto - Anexo III deverá ser protocolizado na Anvisa, sendo dispensada a apresentação de Formulários de Petição - FP1 e FP2 para o mesmo e poderá ser objeto de auditoria. Art. 9º Nos casos de mudanças múltiplas paralelas, a empresa deverá protocolizar cada mudança individual apresentando documentação única que contemple todas as provas relativas a cada um dos assuntos de petição, suprimindo documentações repetidas. Art. 10 Nos casos em que for solicitado relatório de estudo de estabilidade, poderá ser apresentado o estudo de estabilidade acelerado,obrigatoriamente acompanhado de estudo de estabilidade de longa duração em andamento ou estudo de estabilidade de longa duração concluído. Art. 11 Nos casos em que for exigido protocolo ou relatório de estudo de estabilidade, os dados do estudo de estabilidade gerados após o peticionamento deverão ser incluídos no Histórico de Mudanças do Produto. Art. 12 Os resultados fora de especificação do estudo de estabilidade em andamento devem ser informados imediatamente à Anvisa e, posteriormente à conclusão da investigação, deverá ser enviada proposta de ação corretiva. 39
40 Art. 13 O prazo de validade do medicamento será definido de acordo com os resultados de estabilidade apresentados. Parágrafo único. Nos casos em que o estudo enviado comprovar prazo de validade provisório inferior àquele já registrado, o mesmo será reduzido e não será necessário o peticionamento da redução do prazo de validade. Art. 14 Nos casos em que seja solicitado protocolo de estudo de estabilidade, o prazo de validade registrado será mantido. Art. 15 Os Anexos I, II, IV, V e VI referidos nesta norma devem ser apresentados de acordo com os modelos propostos devidamente assinados pelo responsável técnico da empresa detentora do registro. Art. 16 Para a categoria de medicamentos específicos, não será necessário o envio dos documentos a seguir: I. Relatórios técnicos de estudo de biodisponibilidade relativa/ bioequivalência; e II. Relatório de perfil de dissolução comparativo. Art. 17 Não será necessário anexar à petição os novos modelos de texto de bula e rotulagem para as alterações pós-registro que necessitem de atualização dos mesmos, exceto quando solicitados nesta norma ou a critério da Anvisa. 1º A empresa deverá atualizar as informações na bula somente após a aprovação das adequações, alterações, exclusões, inclusões ou reduções pós-registro. 2º A empresa deverá atualizar as informações na bula e rotulagem referentes aos itens I, II, VII, IX e X do art. 5º imediatamente após a alteração, observadas outras regras específicas. Art. 18 Nos casos em que a solicitação pós-registro se referir a mais de uma concentração de uma mesma forma farmacêutica, a mesma deverá ser protocolizada com relatório de estabilidade, relatório de produção e laudo de controle de qualidade referente à maior e menor concentração. Capítulo III DAS MUDANÇAS RELACIONADAS AO LOCAL DE FABRICAÇÃO Seção I Da Alteração ou Inclusão de local de embalagem secundária Art. 19. Refere-se às mudanças relacionadas à alteração ou inclusão do local da linha de embalagem secundária. Art. 20. A petição de alteração descrita nesta seção deverá estar acompanhada do Certificado de Boas Práticas de Fabricação válido. Art. 21. As alterações ou inclusões de local de embalagem secundária podem ser implementadas imediatamente após a data de protocolização da petição. Seção II 40
41 Da Alteração ou Inclusão de local de embalagem primária Art. 22. Refere-se às mudanças relacionadas à alteração ou inclusão do local da linha de embalagem primária. Art. 23. É permitida a inclusão ou alteração concomitante de equipamentos da linha de embalagem primária. Art. 24. É permitida a alteração ou inclusão concomitante de local de embalagem secundária, quando se tratar do mesmo local de embalagem primária. Art. 25. A petição de alteração descrita nesta seção deverá estar acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido; II. Protocolo de estudo de estabilidade referente ao 1º lote ou relatório de estudo de estabilidade referente a 1(um) lote. Art. 26. As alterações ou inclusões de local de embalagem primária podem ser implementadas imediatamente após a data de protocolização da petição. Art. 27. As disposições desta seção não se aplicam aos medicamentos estéreis. Seção III Da Alteração ou Inclusão de local de fabricação do medicamento de liberação convencional Art. 28. Refere-se à alteração ou à inclusão de local relacionada a uma ou mais etapas ou a totalidade do processo de fabricação de medicamentos de liberação convencional. 1º Para produtos estéreis, considera-se alteração ou inclusão de local de fabricação do medicamento a substituição ou adição de local de fabrico da linha completa de produção, podendo excetuar-se apenas a etapa de embalagem secundária. 2º As alterações ou inclusões de local de embalagem primária ou secundária, quando isoladas, devem ser realizadas de acordo com as seções específicas. 3º Para fins deste regulamento não deverão ser peticionadas as alterações ou inclusões das etapas de aquisição de materiais, de pesagem, de rotulagem, de estocagem e de expedição do medicamento. Art. 29. É permitida, concomitantemente, a alteração menor ou moderada do processo de produção ou a alteração dos equipamentos. Art. 30. As petições de Alteração ou Inclusão de local de fabricação do medicamento de liberação convencional deverão ser acompanhadas dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; 41
42 IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; VI. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; Parágrafo único. Quando a alteração ou inclusão de local de fabricação de medicamento de liberação convencional não resultar em alteração de processo produtivo e de equipamentos, ou resultar na alteração menor de processo produtivo, ou resultar na alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento, o item V poderá ser substituído por "Protocolo de estudo de estabilidade referente aos 3(três) lotes iniciais". Art. 31. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 30, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 32. As alterações ou inclusões de local de fabricação do medicamento de liberação convencional só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outrasregras específicas para esta petição. Seção IV Da Alteração ou Inclusão de local de fabricação do medicamento de liberação modificada Art. 33. Refere-se à alteração ou à inclusão de local relacionada a uma ou mais etapas ou a totalidade do processo de fabricação, de medicamentos de liberação modificada. 1º Para produtos estéreis, considera-se alteração ou inclusão de local de fabricação do medicamento a substituição ou adição de local de fabrico da linha completa de produção, podendo excetuar-se apenas a etapa de embalagem secundária. 2º As alterações ou inclusões de local de embalagem primária ou secundária, quando isoladas, devem ser realizadas de acordo com as seções específicas. 3º Para fins deste regulamento não deverão ser peticionadas as alterações ou inclusões das etapas de aquisição de materiais, de pesagem, de rotulagem, de estocagem e de expedição do medicamento. 42
43 Art. 34. É permitida, concomitantemente, a alteração do processo de produção ou a alteração dos equipamentos. Art. 35. As petições de alterações ou inclusões de local de fabricação de medicamentos de liberação modificada deverão ser acompanhadas dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; VI. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; VII. Relatório técnico de estudo de biodisponibilidade relativa/ bioequivalência; 1º Quando a alteração ou inclusão de local de fabricação de medicamento de liberação modificada não resultar em alteração de processo produtivo e de equipamentos, ou resultar na alteração menor de processo produtivo, ou resultar na alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento, o item V poderá ser substituído por "Protocolo de estudo de estabilidade referente aos 3(três) lotes iniciais"; 2º Quando a alteração ou inclusão de local de fabricação de medicamento de liberação modificada não resultar em alteração de processo produtivo e de equipamentos, ou resultar na alteração menor de processo produtivo, ou resultar na alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento, é dispensada a apresentação do item VII. Art. 36. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 35, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; 43
44 Art. 37. As alterações ou inclusões de local de fabricação do medicamento de liberação modificada só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo IV DAS MUDANÇAS RELACIONADAS AO PROCESSO DE PRODUÇÃO Art. 38. Refere-se aos ajustes ou alterações no processo de produção do produto acabado. Seção I Da alteração ou inclusão menor do processo de produção Art. 39. Considera-se alteração ou inclusão menor do processo de produção os ajustes de menor impacto no processo produtivo relacionados à alteração de parâmetros de etapas do processo, tais como: velocidade, temperatura, tempo e ordem de adição dos componentes da fórmula. Parágrafo único. A disposição deste artigo não se aplica às mudanças no processo de esterilização. Art. 40. As alterações ou inclusões menores do processo de produção devem ser acompanhadas dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Protocolo de estudo de estabilidade referente ao 1º lote ou relatório de estudo de estabilidade referente a 1(um) lote. Art. 41. As alterações ou inclusões menores do processo de produção podem ser implementadas imediatamente, não necessitando de protocolização e de análise prévia pela Anvisa. Parágrafo único. A documentação exigida deverá ser anexada ao Histórico de Mudanças do Produto. Seção II Da alteração ou inclusão moderada do processo de produção Art. 42. Considera-se alteração ou inclusão moderada do processo de produção os ajustes de impacto moderado no processo produtivo, que não se enquadrem em alteração menor ou maior do processo de produção. 44
45 Parágrafo único. A disposição deste artigo aplica-se às mudanças no processo de esterilização. Art. 43. As petições de alterações ou inclusões moderadas do processo de produção devem ser acompanhadas dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; Art. 44. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 43, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 45. As alterações ou inclusões moderadas do processo de produção só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção III Da alteração ou inclusão maior do processo de produção Art. 46. Considera-se alteração ou inclusão maior do processo de produção as mudanças que altere o tipo de processo de produção, como a mudança de via seca para úmida e vice-versa, ou alterações de produção que impactem no sistema de liberação do fármaco. Art. 47. As petições de alterações ou inclusões maiores do processo de produção devem ser acompanhadas dos seguintes documentos: 45
46 I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; VI. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; VII. Relatório técnico de estudo de biodisponibilidade relativa/ bioequivalência; Art. 48. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 47, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 49. As alterações ou inclusões maiores do processo de produção só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo V DAS MUDANÇAS RELACIONADAS AO EQUIPAMENTO Art. 50. Refere-se à alteração ou inclusão de equipamento com igual ou diferente capacidade, desenho ou princípio de funcionamento ou automatização do equipamento. Seção I Da alteração ou inclusão de equipamento de embalagem primária Art. 51. Refere-se à alteração ou inclusão de equipamento de embalagem primária. 46
47 Art. 52. As alterações ou inclusões de equipamento de embalagem primária podem ser implementadas imediatamente, não necessitando de protocolização e de análise prévia pela Anvisa. Parágrafo único. A referida mudança deverá ser registrada no Histórico de Mudanças do Produto. Seção II Da alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento Art. 53. Refere-se à alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento, excetuando os equipamentos da linha de embalagem. Art. 54. É permitida a variação da capacidade, a automatização do equipamento ou alteração menor do processo de produção concomitantemente com a alteração a que se refere esta seção. Art. 55. As alterações ou inclusões de equipamento com mesmo desenho e princípio de funcionamento devem ser acompanhadas dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada;ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Protocolo de estudo de estabilidade referente ao 1º lote ou relatório de estudo de estabilidade referente a 1(um) lote. Parágrafo único. Quando se tratar de inclusão de equipamento com mesma capacidade, sistema de automatização e processo produtivo é dispensada a apresentação do item IV e V. Art. 56. As alterações ou inclusões de equipamento com mesmo desenho e princípio de funcionamento podem ser implementadas imediatamente, não necessitando de protocolização e análise prévia pela Anvisa. Parágrafo único. A documentação exigida deverá ser anexada ao Histórico de Mudanças do Produto. Seção III Da alteração ou inclusão de equipamento com diferente desenho ou princípio de funcionamento 47
48 Art. 57. Refere-se à alteração ou inclusão de equipamento com diferente desenho e princípio de funcionamento ou de equipamento com diferente desenho e mesmo princípio de funcionamento,excetuando os equipamentos da linha de embalagem. Art. 58. É permitida, concomitantemente, a alteração menor e moderada do processo de produção em função da alteração de equipamento. Art. 59. A petição de alteração ou inclusão de equipamento com diferente desenho e princípio de funcionamento ou de equipamento com diferente desenho e mesmo princípio de funcionamentodeve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada;ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; VI. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; Art. 60. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 59, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 61. As alterações ou inclusões de equipamento com diferente desenho ou princípio de funcionamento só poderão ser implementadas após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo VI 48
49 DAS MUDANÇAS RELACIONADAS AO TAMANHO DO LOTE Da inclusão de tamanho de lote em até 10 vezes Seção I Art. 62. Refere-se à inclusão de tamanho de lote em até 10 vezes o tamanho do lote piloto/biolote. Parágrafo único. A disposição deste artigo não se aplica aos medicamentos de concentração inferior a 0,99mg por unidade posológica, exceto para soluções perfeitas. Art. 63. É permitida, concomitantemente, a alteração menor do processo de produção e alteração ou inclusão de equipamento com mesmo desenho e mesmo princípio de funcionamento, podendo variar a capacidade e/ou automatização do equipamento. Art. 64. A inclusão de tamanho de lote em até 10 vezes deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a","c" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Protocolo de estudo de estabilidade referente ao 1º lote ou relatório de estudo de estabilidade referente a 1(um) lote. Art. 65. A inclusão de tamanho de lote em até 10 vezes pode ser implementada imediatamente, não necessitando de protocolização e análise prévia pela Anvisa. Parágrafo único. A documentação exigida deverá ser anexada ao Histórico de Mudanças do Produto. Seção II Da inclusão de tamanho do lote superior a 10 vezes Art. 66. Refere-se à inclusão de tamanho de lote superior a 10 vezes o tamanho do lote piloto/biolote. Art. 67. Aplica-se a qualquer inclusão de tamanho de lote para medicamentos de concentração inferior a 0,99 mg por unidade posológica, exceto para soluções perfeitas 49
50 Art. 68. É permitida, concomitantemente, a alteração menor do processo de produção e alteração ou inclusão de equipamento com mesmo desenho e mesmo princípio de funcionamento, podendo variar a capacidade e/ou automatização do equipamento. Art. 69. A petição de inclusão do tamanho do lote deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a","c" e "d" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; Art. 70. A inclusão de tamanho do lote superior a 10 vezes o tamanho do lote piloto/biolote só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo VII DAS MUDANÇAS RELACIONADAS AOS EXCIPIENTES Art. 71. Refere-se às mudanças quantitativas e qualitativas do(s) excipiente(s) da formulação Seção I Da inclusão de nova apresentação por alteração de sabor Art. 72. Refere-se à inclusão de sabor através da adição ou exclusão de aromatizante, edulcorante, flavorizante ou corante a uma formulação já registrada. Art. 73. A presente inclusão resulta em novo número de registro e não cancela o anterior. 1º Caso não exista interesse em manter a apresentação anterior, deverá ser peticionado o cancelamento de apresentação. 2º Caso a empresa queira alterar sabor, de acordo com o disposto no Art. 72, sem resultar em um novo número de registro, deverá ser peticionada a alteração menor ou moderada de excipientes. Art. 74. A petição de inclusão de sabor, odor ou cor deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a 50
51 inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Código GTIN para a(s) nova(s) apresentação (ões); III. Relatório de produção, incluindo os quadros comparativos "a" e "b" do Anexo V; IV. Especificação e metodologia analítica, com referência bibliográfica utilizada, ou, quando não farmacopeica, descrição da metodologia para os excipientes cujas informações ainda não constem no registro. V. Informações referentes à encefalopatia espongiforme transmissível, para os excipientes cujas informações ainda não constem no registro; VI. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VII. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; VIII. Relatório de validação do novo método analítico do produto acabado; IX. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; Parágrafo único. Nos casos em que a solicitação resultar em exclusão de corante, edulcorante, flavorizante e/ou aromatizante a uma formulação já registrada, permite-se a apresentação do protocolo de estabilidade do 1 lote em substituição ao relatório de estudo de estabilidade de 1(um) lote Art. 75. A inclusão de nova apresentação por alteração de sabor só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da alteração menor de excipiente Art. 76. Refere-se à redução ou exclusão de corante, edulcorante, flavorizante ou aromatizante e às alterações quantitativas que se enquadrarem nos limites descritos no Anexo de excipientes Anexo II. Art. 77. A petição de alteração menor de excipiente deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "b" do Anexo V; III. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; 51
52 IV. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; V. Relatório de validação do novo método analítico do produto acabado; VI. Protocolo de estudo de estabilidade referente a 1(um) lote do produto acabado; VII. Relatório com método e resultados dos testes de eficácia de conservantes, nos casos em que se altera o próprio sistema conservante; Parágrafo único. Quando se tratar de redução ou exclusão de excipientes relativos à cor sabor ou odor será dispensada a apresentação do item V. Art. 78. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 77, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 79. A alteração menor de excipiente pode ser implementada imediatamente após a protocolização da petição. Seção III Da alteração moderada de excipiente Art. 80. Refere-se às mudanças quantitativas e qualitativas de excipientes que se enquadrarem nos limites descritos no Anexo de excipientes - Anexo II e às alterações referentes às formas farmacêuticas não contempladas pelo referido Anexo. Art. 81. A petição de alteração moderada de excipiente deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresentesituação satisfatória de acordo com a última inspeção realizada;ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "b" do Anexo V; III. Especificação e metodologia analítica, com referência bibliográfica utilizada, ou, quando não farmacopeica, descrição da metodologia para os excipientes cujas informações ainda não constem no registro. 52
53 IV. Informações referentes à encefalopatia espongiforme transmissível, para os excipientes cujas informações ainda não constem no registro; V. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VI. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; VII. Relatório de validação do novo método analítico do produto acabado; VIII. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; IX. Relatório com método e resultados dos testes de eficácia de conservantes, nos casos em que se altera o próprio sistema conservante; Art. 82. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 81, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 83. A alteração moderada de excipiente só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção IV Da alteração maior de excipiente Art. 84 Refere-se às mudanças quantitativas e qualitativas que estiverem acima dos limites descritos para alteração moderada do Anexo de excipientes - Anexo II. Art. 85. A petição de alteração maior de excipiente deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "b" do Anexo V; 53
54 III. Especificação e metodologia analítica, com referência bibliográfica utilizada, ou, quando não farmacopeica, descrição da metodologia para os excipientes cujas informações ainda não constem no registro. IV. Informações referentes à encefalopatia espongiforme transmissível, para os excipientes cujas informações ainda não constem no registro; V. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VI. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; VII. Relatório de validação do novo método analítico do produto acabado; VIII. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; IX. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; X. Relatório com método e resultados dos testes de eficácia de conservantes, nos casos em que se altera o próprio sistema conservante; XI. Relatório técnico de estudo de biodisponibilidade relativa/ bioequivalência; Art. 86. Para produtos semi-sólidos e líquidos, excetuando-se as soluções perfeitas, aplicam-se, além das contidas no art. 85, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art. 87. A alteração maior de excipiente só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo VIII DAS MUDANÇAS RELACIONADAS À ATUALIZAÇÃO DE ESPECIFICAÇÕES E MÉTODOS ANALÍTICOS DO PRODUTO ACABADO Art. 88. Refere-se à alteração, inclusão ou exclusão de método e/ou especificação do produto acabado que não seja decorrente de alteração pós-registro. 54
55 Parágrafo único. A alteração, inclusão ou exclusão de método e/ou especificação do produto acabado que seja decorrente de alteração pós-registro será analisada conjuntamente à alteração proposta. Seção I Da adequação de especificações e métodos analíticos a compêndio oficial ou estreitamento de faixa de especificação Art. 89. Refere-se à mudança da faixa de especificação e à atualização, inclusão ou substituição do método analítico para fins de adequação a compêndio oficial, ou ainda a qualquer estreitamento da faixa de especificação. Parágrafo único. A disposição deste artigo não se aplica a atualização/inclusão/substituição de método analítico referente a produtos de degradação e método biológico de quantificação de teor. Art. 90. A adequação de especificações e métodos analíticos a compêndio oficial ou estreitamento de faixa de especificação deve ser acompanhada da descrição da especificação ou método analítico já aprovado e do alterado, incluindo a nova referência. Art. 91. A adequação de especificações e método analítico a compêndio oficial ou estreitamento de faixa de especificação pode ser implementada imediatamente, não necessitando de protocolização e análise prévia pela Anvisa. Parágrafo único. A documentação exigida deverá ser anexada ao Histórico de Mudanças do Produto. Seção II Da atualização de especificações e métodos analíticos Art. 92. Refere-se à: a. Atualização de especificações e de método(s) analítico(s) nos casos em que ocorra alteração ou inclusão de método(s) analítico( s) ou de especificação(ões) que não conste nos compêndios oficiais aceitos pela Anvisa; b. Atualização ou substituição ou inclusão de método(s) analítico(s) ou especificação(ões) de produtos de degradação ou de método(s) biológico(s) de quantificação de teor; c. Exclusão de método(s) analítico(s) ou especificação(ões). Art. 93. A petição de atualização de especificações e método analítico deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Descrição da especificação ou método analítico já aprovado e do alterado, incluindo a nova referência; 55
56 III. Referências bibliográficas e/ou cópia de compêndio; IV. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; V. Relatório de validação do novo método analítico do produto acabado; Art. 94. Não é permitida exclusão de ensaio, método analítico ou especificações obrigatórias para a forma farmacêutica. Art. 95. A atualização de especificações e metodologia analítica só pode ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo IX DAS MUDANÇAS RELACIONADAS AO PRAZO DE VALIDADE OU AOS CUIDADOS DE CONSERVAÇÃO Art. 96. Refere-se à alteração do prazo de validade ou alteração dos cuidados de conservação do produto acabado, do produto após a reconstituição ou do produto após diluição. Seção I Da redução do prazo de validade com manutenção dos cuidados de conservação Art. 97. Refere-se à redução do prazo de validade do produto acabado, do produto após a reconstituição ou do produto após diluição mantendo-se os cuidados de conservação inalterados. Art. 98. A petição de redução do prazo de validade com manutenção dos cuidados de conservação deve ser acompanhada do relatório de estudo de estabilidade referente a 1(um) lote de longa duração ou acompanhamento. Art. 99. A redução do prazo de validade, mantendo os cuidados de conservação inalterados, pode ser implementada imediatamente após a protocolização da petição, não necessitando de análise prévia pela Anvisa. Seção II Da redução do prazo de validade com alteração dos cuidados de conservação Art Refere-se à redução do prazo de validade do produto acabado, do produto após a reconstituição ou do produto após diluição alterando-se os cuidados de conservação Art A petição de redução do prazo de validade com alteração dos cuidados de conservação deve ser acompanhada do relatório de estudo de estabilidade de longa duração referente a 3(três) lotes; Art A redução do prazo de validade com alteração dos cuidados de conservação só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção III 56
57 Da ampliação do prazo de validade ou alteração dos cuidados de conservação Art Refere-se à ampliação do prazo de validade ou alteração dos cuidados de conservação do produto acabado, do produto após a reconstituição ou do produto após diluição. Art A ampliação do prazo de validade ou alteração dos cuidados de conservação deve ser acompanhada do relatório de estudo de estabilidade de longa duração referente a 3(três) lotes; Art A ampliação do prazo de validade ou alteração dos cuidados de conservação só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo X DA INCLUSÃO DE NOVA APRESENTAÇÃO COMERCIAL Art Refere-se à inclusão de nova apresentação na qual ocorra alteração no volume ou no número de unidades farmacotécnicas previamente registradas, ou inclusão, alteração ou retirada de acessórios. 1º Caso não exista interesse em manter as apresentações anteriores, deverá ser peticionado o cancelamento de apresentação. 2º A nova apresentação deverá ser condizente com a posologia do produto. 3º Para a inclusão de nova apresentação fracionável aplicase, além do disposto neste capítulo, o disposto em normativa específica. Seção I Da inclusão de nova apresentação comercial Art Refere-se à inclusão de nova apresentação comercial de produto não estéril e a todos os casos em que ocorra inclusão, alteração ou retirada de acessórios. Art A petição de inclusão de nova apresentação comercial de produto não estéril deve ser acompanhada de código GTIN para a(s) nova(s) apresentação(ões); Parágrafo único. Para produtos líquidos em que a nova apresentação sofra alteração do volume deve ser apresentado protocolo de estudo de estabilidade referente ao 1º lote ou relatório de estudo de estabilidade referente a 1(um) lote. Art A inclusão de nova apresentação comercial de produto não estéril e de todos os casos em que ocorra inclusão, alteração ou retirada de acessórios só poderão ser implementados após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da inclusão de nova apresentação comercial de produto estéril Art Refere-se à inclusão de nova apresentação comercial de produto estéril. 57
58 Art A petição de inclusão de nova apresentação comercial de produto estéril deve ser acompanhada de código GTIN para a(s) nova(s) apresentação(ões); Parágrafo único. Para produtos líquidos estéreis em que a nova apresentação sofra alteração do volume deve ser apresentado relatório de estudo de estabilidade referente a 1(um) lote. Art A inclusão de nova apresentação comercial de produto estéril só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XI DA INCLUSÃO DE NOVO ACONDICIONAMENTO Art Refere-se à inclusão de um novo acondicionamento ou de acondicionamento fracionável para um produto já registrado. Parágrafo único. Caso não exista interesse em manter o acondicionamento anterior, a detentora deverá solicitar o cancelamento de registro da(s) apresentação(ões) na justificativa técnica. Art As apresentações resultantes da inclusão de novo acondicionamento fracionável devem atender, além do disposto neste capítulo, o que dispõe a normativa específica. Art É permitida, concomitantemente, a alteração dos equipamentos utilizados exclusivamente para o processo de embalagem. Art A petição de inclusão de novo acondicionamento deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de estudo de estabilidade referente a 3(três) lotes; III. Relatório com método e resultados do controle de qualidade de embalagem para soluções parenterais de pequeno e grande volume e nutrição parenteral; IV. Especificação do material de acondicionamento; V. Código GTIN para a(s) nova(s) apresentação(ões). Art Nos casos de inclusão de um novo acondicionamento que atenda as condições descritas no Anexo VII Materiais de acondicionamento, o item II do Art. 116 poderá ser substituído pelo protocolo de estudo de estabilidade referente aos 3(três) lotes iniciais. Parágrafo único. A disposição deste artigo não se aplica aos medicamentos injetáveis. 58
59 Art Nos casos de inclusão de um novo acondicionamento que atenda as condições descritas no Anexo VII Materiais de acondicionamento, serão mantidos para o novo acondicionamento o prazo de validade e os cuidados de conservação do acondicionamento já registrado. Art A inclusão de novo acondicionamento só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XII DAS MUDANÇAS RELACIONADAS AO FÁRMACO Art Refere-se à alteração ou inclusão na rota de síntese do fármaco ou de local de fabricação do fármaco. Parágrafo único. A disposição deste capítulo não se aplica à categoria de medicamentos específicos. Art Toda a documentação emitida pelo fabricante do fármaco deverá ser enviada em papel timbrado. Parágrafo Único. Fica facultado ao(s) fabricante(s) do(s) Fármaco(s) enviar(em) diretamente à Anvisa a documentação relativa ao fármaco, devidamente identificada com o nome da empresa detentora do registro, o número do processo e o número do expediente a que se relaciona. Seção I Da alteração ou inclusão da rota de síntese do fármaco Art Refere-se à alteração ou inclusão na rota de síntese do fármaco, permanecendo o mesmo fabricante já informado no registro. Art A petição de alteração ou inclusão da rota de síntese do fármaco deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Perfil comparativo de impurezas entre 1(um) lote do fármaco obtido pela rota de síntese aprovada no registro e 1(um) lote do fármaco obtido pela nova rota de síntese; III. Relatório de estudo de estabilidade de 1(um) lote do fármaco; IV. Rota de síntese completa com produtos intermediários; V. Laudo analítico de controle de qualidade do fármaco referente a 1(um) lote emitido pelo fabricante do medicamento; 59
60 VI. Laudo analítico de controle de qualidade do fármaco referente a 1(um) lote emitido pelo fabricante do fármaco; VII. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VIII. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; IX. Relatório de validação do novo método analítico do produto acabado; X. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; 1º A rota de síntese deverá conter as informações referentes aos solventes utilizados, lista de solventes residuais, polimorfismo, aos limites, quantificação e especificação de impurezas de síntese e produtos de degradação, além das informações referentes à quiralidade e proporção de isômeros. 2º Deverá ser apresentada discussão relativa ao impacto de eventuais alterações do perfil de impureza e validação, ou revalidação, da metodologia de análise. Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 123, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; Art A alteração ou inclusão da rota de síntese do fármaco só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da alteração ou inclusão de local de fabricação do fármaco Art Refere-se à substituição ou adição do local de fabricação do fármaco. Art É permitida, concomitantemente, a alteração ou inclusão da rota de síntese do fármaco em função da alteração ou inclusão do local de fabricação do fármaco. Art A petição de alteração ou inclusão de local de fabricação do fármaco deve ser acompanhada dos seguintes documentos: 60
61 I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Perfil comparativo de impurezas entre 1(um) lote do fármaco obtido pela rota de síntese e/ou local de fabricação aprovada no registro e 1(um) lote do fármaco obtido pela nova rota de síntese e/ou local de fabricação; III. Relatório de estudo de estabilidade de 1(um) lote do fármaco; IV. Rota de síntese completa com produtos intermediários; V. Laudo analítico de controle de qualidade do fármaco referente a 1(um) lote emitido pelo fabricante do medicamento; VI. Laudo analítico de controle de qualidade do fármaco referente a 1(um) lote emitido pelo fabricante do fármaco; VII. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VIII. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; IX. Relatório de validação do novo método analítico do produto acabado; X. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; 1º A rota de síntese deverá conter as informações referentes aos solventes utilizados, lista de solventes residuais, polimorfismo, aos limites, quantificação e especificação de impurezas de síntese e produtos de degradação, além das informações referentes à quiralidade e proporção de isômeros. 2º Deverá ser apresentada discussão relativa ao impacto de eventuais alterações do perfil de impureza e validação, ou revalidação, do método de análise para o fármaco. Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 128, as seguintes regras: I. Apresentar resultados comparativos entre distribuição do tamanho de partícula/gotícula da condição anteriormente registrada e da nova condição; II. Incluir discussão relativa ao impacto de eventuais alterações da distribuição do tamanho de partícula/gotícula; III. Apresentar resultados comparativos entre a taxa de permeação cutânea da condição anteriormente registrada e da nova condição. IV. Incluir discussão relativa ao impacto de eventuais alterações da taxa de permeação cutânea; 61
62 Art A alteração ou inclusão de local de fabricação do fármaco só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XIII DAS MUDANÇAS RELACIONADAS À POSOLOGIA Art Refere-se à alteração de posologia para um produto já registrado de mesma concentração, forma farmacêutica e indicação terapêutica. Da alteração de posologia Seção I Art Refere-se aos casos de alteração de posologia no registro de medicamentos novos e de referência. Art A petição de alteração de posologia deve ser acompanhada dos seguintes documentos: I. Relatório de estudo clínico fase III. II. Texto de bula atualizado Art A alteração de posologia só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da alteração de posologia para medicamentos específicos Art Refere-se as mudanças de posologia no registro de medicamentos específicos. Art A petição de alteração de posologia para medicamentos específicos deve ser acompanhada dos seguintes documentos: I. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas, para posologia que não se enquadre dentro dos limites já estabelecidos em normas vigentes; II. Texto de bula atualizado. Art A alteração de posologia para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XIV DA AMPLIAÇÃO DE USO Art Refere-se ao aumento da população alvo para um produto já registrado na mesma indicação terapêutica. 62
63 Seção I Da ampliação de uso Art Refere-se à ampliação de uso no registro de medicamentos novos e de referência. Art A petição de ampliação de uso deve ser acompanhada dos seguintes documentos: I. Relatório de estudo clínico fase III ou IV II. Texto de bula atualizado Art A ampliação de uso só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da ampliação de uso para medicamentos específicos Art Refere-se à ampliação de uso no registro da categoria de medicamentos específicos. Art A petição de ampliação de uso deve ser acompanhada dos seguintes documentos: I. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas II. Texto de bula atualizado Art A ampliação de uso para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XV DA INCLUSÃO DE NOVA VIA DE ADMINISTRAÇÃO Art Refere-se à inclusão de nova via de administração para um produto já registrado na mesma concentração, indicação terapêutica e forma farmacêutica. Seção I Da inclusão de nova via de administração no país Art Refere-se a medicamentos novos e de referência. Art A petição de inclusão de nova via de administração no país deve ser acompanhada dos seguintes documentos I. Relatório de estudo clínico fase II e III II. Texto de bula atualizado 63
64 Art A inclusão de nova via de administração no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da inclusão de nova via de administração já registrada no país Art Refere-se a medicamentos genéricos e similares que incluam nova via de administração já aprovada para o medicamento de referência. Art A petição de inclusão de nova via de administração já registrada no país deve ser acompanhada dos seguintes documentos: I. Relatório de equivalência farmacêutica; II. Relatório de perfil de dissolução comparativo, quando aplicável III. Relatório técnico de estudo de biodisponibilidade relativa/bioequivalência; Art A inclusão de nova via de administração já registrada no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção III Da inclusão de nova via de administração para medicamentos específicos Art Refere-se à categoria de medicamentos específicos. Art A petição de inclusão de nova via de administração para medicamentos específicos deve ser acompanhada dos seguintes documentos: I. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas, para via de administração nova no país; II. Texto de bula atualizado. Art A inclusão de nova via de administração para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XVI DA INCLUSÃO DE INDICAÇÃO TERAPÊUTICA Art Refere-se à inclusão de indicação terapêutica para um produto já registrado na mesma concentração e forma farmacêutica. Seção I Da inclusão de indicação terapêutica nova no país 64
65 Art Refere-se a medicamentos novos e de referência. Art A petição de inclusão de indicação terapêutica nova no país deve ser acompanhada dos seguintes documentos I. Relatório de estudo clínico fase II e III; II. Texto de bula atualizado. Art A inclusão de indicação terapêutica nova no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção III Da inclusão de indicação terapêutica para medicamentos específicos Art Refere-se à categoria de medicamentos específicos. Art A petição de inclusão de indicação terapêutica para medicamentos específicos deve ser acompanhada dos seguintes documentos: I. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas; II. Texto de bula atualizada. Art A inclusão de indicação terapêutica para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XVII DA INCLUSÃO DE NOVA CONCENTRAÇÃO Art Refere-se à inclusão de nova concentração para um produto já registrado na mesma forma farmacêutica e indicação terapêutica. Seção I Da Inclusão de nova concentração no país Art Refere-se a medicamentos novos e de referência. Art A petição de inclusão de nova concentração no país deve ser acompanhada dos relatórios técnicos e toda a documentação de acordo com as normas vigentes sobre o registro de medicamentos novos. Art A inclusão de nova concentração no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II 65
66 Da inclusão de nova concentração já registrada no país Art Refere-se a medicamentos genéricos, novos e similares. Art Para medicamentos genéricos e similares a petição de inclusão de nova concentração já registrada no país deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Código GTIN para a(s) nova(s) apresentação(ões); III. Relatório de produção; IV. Laudo analítico de controle de qualidade do fármaco e do produto acabado referente a 1 (um) lote incluindo especificações e referências bibliográficas (caso haja alteração de metodologia analítica, anexar respectiva validação); V. Relatório de equivalência farmacêutica; VI. Relatório de perfil de dissolução comparativo, quando aplicável VII. Especificação do material de acondicionamento; VIII. Relatório com método e resultados do controle de qualidade de embalagem para soluções parenterais de pequeno e grande volume e nutrição parenteral; IX. Relatório de estudo de estabilidade referente a 3(três) lotes; XI. Relatório com método e resultados dos testes de eficácia de conservantes, quando aplicável XII. Relatório técnico de estudo de biodisponibilidade relativa/bioequivalência; Art Para medicamentos novos, a petição de inclusão de nova concentração já registrada no país deve ser acompanhada dos seguintes documentos de relatórios técnicos e toda a documentação de acordo com as normas vigentes sobre o registro de medicamentos novos. Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 167, as seguintes regras: I. Determinar, com metodologia adequada, a distribuição do tamanho de partícula/gotícula; II. Determinar, com metodologia adequada, a taxa de permeação cutânea. Art A inclusão de nova concentração já registrada no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. 66
67 Seção III Da inclusão de nova concentração para medicamentos específicos Art Refere-se à categoria medicamentos específicos. Art A petição de inclusão de nova concentração para medicamentos específicos deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Código GTIN para a(s) nova(s) apresentação(ões); III. Relatório de produção; IV. Laudo analítico de controle de qualidade do fármaco e do produto acabado referente a 1 (um) lote incluindo especificações e referências bibliográficas (caso haja alteração de metodologia analítica, anexar respectiva validação); V. Especificação do material de acondicionamento; VI. Relatório com método e resultados do controle de qualidade de embalagem para soluções parenterais de pequeno e grande volume e nutrição parenteral; VII. Relatório de estudo de estabilidade referente a 3(três) lotes; VIII. Relatório com método e resultados dos testes de eficácia de conservantes, quando aplicável IX. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas, para concentrações novas no país ou para concentrações que não se enquadram dentro dos limites já estabelecidos em normas vigentes. X. Texto de bula atualizado Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 172, as seguintes regras: I. Determinar, com metodologia adequada, a distribuição do tamanho de partícula/gotícula; II. Determinar, com metodologia adequada, a taxa de permeação cutânea. Art A inclusão de nova concentração para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XVIII 67
68 DA INCLUSÃO DE NOVA FORMA FARMACÊUTICA Art Refere-se à inclusão de nova forma farmacêutica para um produto já registrado na mesma indicação terapêutica. Seção I Da inclusão de nova forma farmacêutica no país Art Refere-se a medicamentos novos e de referência. Art A petição de inclusão de nova forma farmacêutica no país deve ser acompanhada dos relatórios técnicos e de toda a documentação nos termos da norma vigente sobre o registro de medicamentos novos. Art A inclusão de nova forma farmacêutica no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção II Da inclusão de nova forma farmacêutica já registrada no país Art Refere-se a medicamentos similares e novos. Art Para medicamentos genéricos e similares a petição de inclusão de nova forma farmacêutica já registrada no país deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Código GTIN para a(s) nova(s) apresentação(ões); III. Relatório de produção; IV. Laudo analítico de controle de qualidade do fármaco e do produto acabado referente a 1 (um) lote incluindo especificações e referências bibliográficas (caso haja alteração de metodologia analítica, anexar respectiva validação); V. Relatório de equivalência farmacêutica; VI. Relatório de perfil de dissolução comparativo, quando aplicável VII. Especificação do material de acondicionamento; VIII. Relatório com método e resultados do controle de qualidade de embalagem para soluções parenterais de pequeno e grande volume e nutrição parenteral; 68
69 IX. Relatório de estudo de estabilidade referente a 3(três) lotes; X. Relatório com método e resultados dos testes de eficácia de conservantes, quando aplicável; XI. Relatório técnico de estudo de biodisponibilidade relativa/bioequivalência; Art Para medicamentos novos a petição de inclusão de nova forma farmacêutica já registrada no país deve ser acompanhada de relatórios técnicos e toda a documentação de acordo com as normas vigentes sobre o registro de medicamentos novos. Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 180, as seguintes regras: I. Determinar, com metodologia adequada, a distribuição do tamanho de partícula/gotícula; II. Determinar, com metodologia adequada, a taxa de permeação cutânea. Art A inclusão de nova forma farmacêutica já registrada no país só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Seção III Da inclusão de nova forma farmacêutica para medicamentos específicos Art Refere-se a inclusão de nova forma farmacêutica para categoria de medicamentos específicos. Art A petição de inclusão de nova forma farmacêutica para medicamentos específicos deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Código GTIN para a(s) nova(s) apresentação(ões); III. Relatório de produção; IV. Laudo analítico de controle de qualidade do fármaco e do produto acabado referente a 1 (um) lote incluindo especificações e referências bibliográficas (caso haja alteração de metodologia analítica, anexar respectiva validação); V. Especificação do material de acondicionamento; VI. Relatório com método e resultados do controle de qualidade de embalagem para soluções parenterais de pequeno e grande volume e nutrição parenteral; VII. Relatório de estudo de estabilidade referente a 3(três) lotes; 69
70 VIII. Relatório com método e resultados dos testes de eficácia de conservantes, quando aplicável IX. Relatório de estudos clínicos ou estudos clínicos publicados em revistas indexadas, para forma farmacêutica nova no país X. Texto de bula atualizado Art Para produtos semi-sólidos e líquidos, excetuandose as soluções perfeitas, aplicam-se, além das contidas no art. 185, as seguintes regras: I. Determinar, com metodologia adequada, a distribuição do tamanho de partícula/gotícula; II. Determinar, com metodologia adequada, a taxa de permeação cutânea. Art A inclusão de nova forma farmacêutica para medicamentos específicos só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XX DAS MUDANÇAS RELACIONADAS À ROTULAGEM Art Refere-se à alteração da rotulagem dos medicamentos já registrados que não tenha sido contemplada em norma específica ou que não seja decorrente de alteração pós-registro. Art A petição de alteração de rotulagem deve ser acompanhada do novo lay-out de rótulo e embalagem. Art A alteração de rotulagem dos medicamentos já registrados que não tenha sido contemplada em normativa específica ou que não seja decorrente de alteração pós-registro só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XXI DAS MUDANÇAS RELACIONADAS AO NOME COMERCIAL Art Refere-se à alteração do nome comercial de medicamentos já registrados. Art A petição de alteração de nome comercial deverá ser acompanhada de declaração de não comercialização do produto. Art A alteração de nome comercial só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XXII DA SUSPENSÃO TEMPORÁRIA DE FABRICAÇÃO Art Refere-se à suspensão temporária da fabricação de um produto registrado não implicando o cancelamento do seu registro. 70
71 Art A solicitação deverá ser protocolizada com 180 dias de antecedência da paralisação de fabricação, exceto em situações emergenciais. Art A suspensão temporária da fabricação de um produto registrado só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Parágrafo único. A empresa poderá implementar a suspensão caso não haja manifestação da Anvisa no período de 180 dias. Capítulo XXIII DA REATIVAÇÃO DA FABRICAÇÃO DE MEDICAMENTO Art Refere-se à solicitação de retomada da fabricação de um produto já registrado. Art Nos casos em que a suspensão da fabricação foi motivada pelo não atendimento de requerimentos técnicos, a reativação da fabricação ficará condicionada ao cumprimento dos requerimentos que motivaram a suspensão. Art A petição de reativação de fabricação deve ser acompanhada de Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. Art A reativação da fabricação de medicamento só poderá ser implementada após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. Capítulo XXIV DO CANCELAMENTO DO REGISTRO Seção I Do cancelamento de registro da apresentação do medicamento Art Refere-se ao cancelamento do registro de determinadas apresentações do medicamento. Art O cancelamento da apresentação do medicamento só poderá ser implementado após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. O cancelamento só poderá ser solicitado após a aprovação da Suspensão Temporária de Fabricação nos termos do Capitulo XXII, exceto para as seguintes situações: 1º Cancelamentos em que a empresa manterá registro de produto na mesma forma farmacêutica e concentração; 2º Cancelamentos em que as apresentações não foram comercializadas. 71
72 Seção II Do cancelamento de registro do medicamento Art Refere-se ao cancelamento do registro de todas as apresentações do medicamento. Art O cancelamento do registro do medicamento só poderá ser implementado após análise e conclusão favorável da Anvisa, observadas outras regras específicas para esta petição. O cancelamento só poderá ser solicitado após a aprovação da Suspensão Temporária de Fabricação nos termos do Capitulo XXII exceto para os medicamentos que não foram comercializados. Capítulo XXV EXCLUSÃO DE LOCAL DE FABRICAÇÃO DO FÁRMACO OU LOCAL DE EMBALAGEM PRIMÁRIA OU LOCAL DE EMBALAGEM SECUNDÁRIA OU LOCAL DE FABRICAÇÃO DO PRODUTO Art As petições de exclusão referidas neste capítulo devem ser acompanhadas da lista dos locais que permanecem vigentes, assinada pelo responsável técnico do detentor do registro. Art As exclusões referidas neste capítulo podem ser implementadas imediatamente após a protocolização da petição. Capítulo XXVI DAS DISPOSIÇÕES FINAIS E TRANSITÓRIAS Art A obrigatoriedade da apresentação de documentos relacionados à determinação da distribuição do tamanho de partícula/gotícula se iniciará no prazo de 180 (cento e oitenta) dias, contados a partir da data da publicação desta norma. Art A obrigatoriedade da apresentação de documentos relacionados à determinação da taxa de permeação cutânea nos termos deste regulamento, se iniciará no prazo a ser determinado em norma específica. Art As decisões da Anvisa quanto à avaliação das solicitações pós-registro serão objeto de publicação em Diário Oficial da União, ou em outro meio de divulgação institucional, quando aplicável. Art Nos casos não previstas nesta Resolução, ou que não satisfaçam a algum dos quesitos especificados, ficará a critério da Anvisa estabelecer os testes e a documentação que deverão ser apresentados. Art Para os medicamentos similares que ainda não cumpriram com os critérios de adequação conforme legislação específica, o relatório técnico de bioequivalência, quando exigido nesta norma, será dispensado até o momento da adequação. Art A Anvisa poderá solicitar documentos adicionais quando julgar necessário. 72
73 Art Recomendações da Anvisa para o pós-registro de medicamentos estarão disponibilizadas para consulta no site desta Agência. Art O descumprimento das disposições contidas nesta Resolução e no regulamento por ela aprovado constitui infração sanitária, nos termos da Lei nº , de 20 de agosto de 1977, sem prejuízo das responsabilidades civil, administrativa e penal cabíveis. Art Ficam revogadas a Resolução - RE nº. 893, de 29 de maio de 2003, a Resolução RE nº. 321, de 13 de setembro de 2004; a Resolução - RE nº. 215, de 24 de junho de 2004; a Resolução - RE nº.1316, de 31 de maio de 2005; a Resolução - RE nº. 2328, de 20 de setembro de 2005; a Instrução Normativa - IN nº. 1, de 21 de março de 2007; a Instrução Normativa - IN nº. 10, de 21 de agosto de 2007; e a Instrução Normativa - IN nº. 3, de 4 de junho de 2008, a Instrução Normativa IN nº. 6, de 25 de maio de Art Esta Resolução entra em vigor no prazo de 5 (cinco) dias, contados a partir da data de sua publicação. DIRCEU RAPOSO DE MELLO ANEXO I JUSTIFICATIVA DA SOLICITAÇÃO Descrição da solicitação1 Razão da solicitação2 Declaro que nenhuma mudança, além da acima proposta, será realizada e que as informações constantes no texto de bula e rotulagem serão alteradas de acordo com a solicitação acima descrita e serão realizadas somente após a aprovação por esta ANVISA Responsável técnico 1. Relato contendo a proposta de alteração solicitada pela empresa 2. Motivação da alteração proposta pela empresa incluindo o argumento técnico para a realização da alteração Quando pertinente a empresa deverá anexar documentação comprobatória da motivação ANEXO II ANEXO DE EXCIPIENTES 1. Determina os critérios para o enquadramento de alterações de excipiente em alteração menor, moderada e maior de excipientes. 73
74 2. Para formas farmacêuticas sólidas de liberação imediata a) Qualquer alteração de excipiente deverá ser baseada na formulação inicialmente registrada ou na última formulação que já tenha segurança e eficácia demonstradas através de estudos clínicos ou bioequivalência, quando aplicável; b) A alteração de cada um dos excipientes e o efeito aditivo total das alterações deve ser calculado considerando alterações de excipientes expressos como porcentagem peso/peso (p/p) do total da formulação. As porcentagens da tabela I estão baseadas na premissa de que o produto foi formulado considerando o princípio ativo com 100% da sua potencia declarada na rotulagem. O peso total da forma farmacêutica deve permanecer dentro da faixa originalmente especificada; Tabela I - Formas farmacêuticas sólidas de liberação imediata Alteração Menor Limite (%) Alteração Moderada Limite (%) Diluente ± 5,0 ± 10,0 Desintegrante 2.1. Amido ± 3,0 ± 6, Outros ± 1,0 ± 2,0 Aglutinante ± 0,5 ± 1,0 Lubrificante 4.1. Estearato de magnésio ou cálcio ± 0,25 ± 0, Outros ± 1,0 ± 2,0 74
75 5. Deslizante 5.1. Talco ± 1,0 ± 2, Outros ± 0,1 ± 0,2 6.Filme de revestimento ± 1,0 ± 2,0 O efeito aditivo das alterações dos excipientes não relacionados ao sistema de liberação modificada do fármaco não pode ser superior a 5%, para alteração menor, e 10 % para alteração moderada. 3. Para formas farmacêuticas sólidas de liberação modificada a) Qualquer alteração de excipiente deverá ser baseada na formulação inicialmente registrada ou na última formulação que já tenha segurança e eficácia demonstradas através de estudos clínicos ou bioequivalência, quando aplicável; b) A alteração de cada um dos excipientes e o efeito aditivo total das alterações deve ser calculado considerando alterações de excipientes expressos como porcentagem peso/peso (p/p) do total da formulação. As porcentagens da tabela I estão baseadas na premissa de que o produto foi formulado considerando o princípio ativo com 100% da sua potencia declarada na rotulagem. O peso total da forma farmacêutica deve permanecer dentro da faixa originalmente especificada; c) A alteração de cada um dos excipientes e o efeito aditivo total das alterações nos excipientes relacionados ao sistema de liberação modificada deve atender o disposto na tabela II, considerando alterações de excipientes expressos como porcentagem peso/peso (p/p) do total da soma dos excipientes que controlam a liberação do fármaco; Tabela I - Formas farmacêuticas sólidas de liberação modificada - Excipientes não relacionados ao sistema de liberação modificada do fármaco. Alteração Menor Limite (%) Alteração Moderada Limite (%) Diluente ± 5,0 ± 10,0 Desintegrante 75
76 2.1. Amido ± 3,0 ± 6, Outros ± 1,0 ± 2,0 Aglutinante ± 0,5 ± 1,0 Lubrificante 4.1. Estearato de magnésio ou cálcio ± 0,25 ± 0, Outros ± 1,0 ± 2,0 Deslizante 5.1. Talco ± 1,0 ± 2, Outros ± 0,1 ± 0,2 Filme de revestimento ± 1,0 ± 2,0 O efeito aditivo das alterações dos excipientes não relacionados ao sistema de liberação modificada do fármaco não pode ser superior a 5%, para alteração menor, e 10 % para alteração moderada. Tabela II - Formas farmacêuticas sólidas de liberação modificada - Excipientes relacionados ao sistema de liberação modificada do fármaco. Alteração Menor Limite (%) Alteração Moderada Limite (%) I - Medicamentos de janela terapêutica estreita ± 5,0 n/a* 76
77 II - Outros ± 5,0 ± 10,0 O efeito aditivo das alterações dos excipientes relacionados ao sistema de liberação modificada do fármaco não pode ser superior a 5%, para alteração menor, e 10 % para alteração moderada. * Para medicamentos de janela terapêutica estreita qualquer alteração acima de 5% nos excipientes relacionados ao sistema de liberação modificada do fármaco constituirá alteração maior de excipientes. Histórico de Mudanças do Produto ANEXO III Cabeçalho Número do processo Nome Comercial Princípio Ativo Formas Farmacêuticas Concentrações Apresentações1 Período 2 Houve alteração? ( ) Sim ( ) Não Pós- Número do expediente e Apresentações envolvidas na Justificativa/descriçAo/ Data da aprovação e Anexo referente a 77
78 Registro data de pmtocolo mudança razão da mudança4 efetivação da mudança mudança 1. Informar todas as apresentações registradas; 2. Período a que se refere o Histórico de Mudanças do Produto no formato: "mm/aaaa a mm/aaaa"; 3. Nome do assunto, segundo a norma vigente, preenchido de acordo com a ordem cronológica da efetivação da mudança; 4. Nos casos em que houve protocolização da mudança informar, neste campo, o respectivo número de expediente e data; 5. A empresa deverá preencher neste campo a justificativa da solicitação contemplando a descrição detalhada e a motivação, incluído o argumento técnico para realização da mudança pós-registro; 6. Informar a data de aprovação e efetivação da mudança proposta. Para solicitações pósregistro de realização imediata informar somente a data da efetivação; 7.Preencher o número do anexo referente aos documentos com os dados gerados em função da mudança de acordo com a norma vigente. O anexo deve conter os seguintes documentos: a. Nos casos em que o pós-registro é reportado apenas no Histórico de Mudanças do Produto deverá ser anexado todos os documentos exigidos pelo assunto; b. Nos casos em que for solicitado protocolo de estabilidade ou for apresentado na solicitação pós-registro estudo de estabilidade acelerado o estudo de estabilidade de longa duração deverá ser anexado quando concluído; ANEXO IV RELATÓRIO DE PRODUÇÃO Cabeçalho Princípio Ativo Nome Comercial Forma Farmacêutica 78
79 Concentração Fórmula mestra Substância Número DCB, DCI ou CAS Quantida de % p/p da forma farmacêut ica Função na Fórmula Informações do lote Tamanho do lote piloto/biolote Tamanho máximo aprovado Tamanho mínimo aprovado Tamanho do lote produzido Ordem de produção Processo produtivo 79
80 Endereço completo (incluindo cidade, país e CNPJ) Lista de equipamentos (incluindo automação, capacidade, desenho e princípio de funcionamento) Descrição do processo farmacotécnico Metodologias de controle em processo(incluindo referencia bibliográfica - Validação) Fluxograma de produção Etapa3 Substanc ia4 Operaç ão Unitári a Parâmetr os da operação unitária4 Equipame nto Control e em process o 1. Enviar cópia da ordem de produção referente ao lote a ser avaliado; 2. Descrever o processo na forma de tópicos numerando cada uma das etapas 3. De acordo com a numeração da descrição do processo farmacotécnico 4. Indicar a ordem de adição das substância na etapa em que esta ocorrer 5. Informações referentes a velocidade, temperatura, tempo, etc. 6. Informar quais os testes que serão realizados e em qual etapa ocorrerão ANEXO V QUADROS COMPARATIVOS 80
81 abeçalho Princípio Ativo Nome Comercial Forma Farmacêutica Quadro B - Comparativo de fórmula Fórmula anterior Fórmula proposta Dife renç as entre as % Substância Número DCB, DCI ou CAS Função Concentraçã o em mg % na fór mu la Concentração em mg % na For mu la Ativo Excipiente 01 Excipiente 02 Excipiente 03 Excipiente 04 Peso médio = Peso médio = das alter a- ções em % = 81
82 Quadro C - Comparativo de tamanho do lote Lote Aprovado Lote Proposto Tamanho do lote piloto/biolote Tamanho máximo Tamanho mínimo Tamanho do lote produzido Quadro D - Comparativo de processo de produção Processo Aprovado Processo Proposto Lista de equipamentos (incluindo automação, capacidade, desenho e princípio de funcionamento) Descrição do processo farmacotécnico Metodologias de controle em processo com especificação 82
83 Fluxograma de produçâo aprovado Etapa2 Substan cia3 Operaçã o Unitária Parâmet ros da operaçã o unitária 4 Equipam entos Controle em processo Fluxograma de produção proposto Etapa2 Substan cia3 Unitária Parâmet ros da operaçã o unitária 4 Equipam entos Controle em processo 7. Descrever o processo na forma de tópicos numerando cada uma das etapas 8. De acordo com a numeração da descrição do processo farmacotécnico 9. Indicar a ordem de adição das substância na etapa em que esta ocorrer 10. Informações referentes a velocidade, temperatura, tempo, etc. 11. Informar quais os testes que serão realizados e em qual etapa ocorrerão ANEXO VI 83
84 RELATÓRIO DE ESTABILIDADE 1. Informações que devem constar no relatório de estabilidade Tipo de Estudo/Condições: Produto: Data de início do estudo: Apresentação: N total de amostras: Especificação da embalagem primária: Fabricante do fármaco: Local de Fabricação/Data de Fabricação: Lote do fármaco: Número do Lote/tamanho do lote: 2. Cronograma SÓLIDOS Especificação Referência do método início 3 meses 6 meses 9 meses 12 meses 18 meses 24 meses 1) Aspecto 1,2 e e 3 2)Teor 12e3 1e2 1e2 2 2e3 2 2e3 3)quantificação produtos degradação 1,2e3 1e2 1e2 2 2e3 2 2e3 84
85 4)Dissolução 1,2e3 1e2 1e2 2 2e3 2 2e3 5) Limites microbianos 1.2 e e 3 6)Dureza 12e e3 LÍQUIDOS E SEMI-SÓLIDOS Especificação Referência do método início 3 meses 6 meses 9 meses 12 meses 18 meses 24 meses 1) Aspecto 1.2 e e 3 2)Teor 12e3 1e2 1e2 2 2e3 2 2e3 3)quantificação produtos degradação 1,2e3 1e2 1e2 2 2e3 2 2e3 4)pH 12e 3 1e2 1e2 2 2e3 2 2e3 5)Sedimentação pos agitação em suspensões 1,2e e3 6)Claridade em soluções 12e e3 7) Separação de fase em emulsões e cremes 1,2 e e 3 85
86 8)Perda de peso em produto de base aquosa 1,2e e3 9) Limites microbianos 1.2 e e Acelerado 2 - Longa 3 - Acompanhamento a. A empresa deverá incluir os testes adicionais necessários para a avaliação do produto; b. Estudo adicionais tais como, fotoestabilidade, validade do produto após reconstituição/diluição e estabilidade após abertura da embalagem, devem ser incluídos no relatório de estabilidade de acordo com o GUIA PARA A REALIZAÇÃO DE ESTUDOS DE ESTABILIDADE; c. Quando algum dos ensaios supracitados não se aplicar ao produto, a empresa deverá enviar justificativa técnica sobre a ausência do mesmo; ANEXO VII MATERIAIS DE ACONDICIONAMENTO Critérios aplicados para a substituição do relatório de estudo de estabilidade por protocolo de estudo de estabilidade para as alterações de um novo acondicionamento - Alteração de material de acondicionamento para frascos de produtos sólidos, semi-sólidos e líquidos não estéreis. Condição Registrada Condição Proposta Poliestiieno PVC Polietileno Polipmpileno 86
87 Vidm PVC Polietileno Polipmpileno Vidm Polietileno Qualquer mudança entre vidro, metal, polipropileno de densidade superior a 0,89 e polietileno de densidade superior a 0,95. Condições específicas: A utilização deste anexo para produtos semi-sólidos e líquidos só será aceita caso sejam de base aquosa e não contenham solventes orgânicos; O material de acondicionamento proposto deve possuir propriedade de barreira a luz equivalente ao que esta sendo comparado ou deve ser apresentado estudo de fotoestabilidade ou justificativa técnica com evidência científica de que os ativos não sofrem degradação na presença de luz ou de que a nova embalagem primária não permite a passagem de luz. - Alteração de material de acondicionamento para blisters de produtos sólidos, semi-sólidos não estéreis. Condição Registrada Condição Proposta PVC PVC/PVDC PVC/PCFFE 87
88 PVC/PVDC/PE PVC/PVDC PVC/PCFFE PVC/PVDC/PE PP PVC/PVDC PVC/PVDC/PE PVC PP Qualquer plástico Blister AL/AL Strip AL/AL O material de acondicionamento proposto deve possuir propriedade de barreira a luz equivalente ao que esta sendo comparado ou deve ser apresentado estudo de fotoestabilidade ou justificativa técnica com evidência científica de que os ativos não sofrem degradação na presença de luz ou de que a nova embalagem primária não permite a passagem de luz. 88
89 RDC Nº 29, DE 17 DE ABRIL DE 2007 Dispõe sobre as regras referentes ao registro e comercialização para a substituição do sistema de infusão aberto para fechado em Soluções Parenterais de Grande Volume. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 354 da ANVISA, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada em 20 de março de 2006, e Considerando as disposições transitórias da RDC n 45/03 que dispõe sobre o regulamento técnico de Boas Praticas de Utilização de Soluções Parenterais em Serviço de Saúde; adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º. Aprovar as regras referentes ao registro e comercialização para a substituição do sistema de infusão aberto para fechado em Soluções Parenterais de Grande Volume. Art 2º. Para a adequação ao sistema fechado, as empresas detentoras de registro de Soluções Parenterais de Grande Volume em Sistema Aberto deverão proceder da seguinte maneira: I - Peticionar com o assunto - "ESPECÍFICO: Inclusão de novo acondicionamento" com o pagamento de taxa correspondente. II - Constar em sua rotulagem os seguintes dizeres "SISTEMA FECHADO". III - Apresentar todos os ensaios de controle de qualidade especificados abaixo: 1. Requisitos Físicos 1.1. Controle Visual Solda de Bico Distribuição de Material Transparência Permeabilidade ao vapor d água Resistência da base do bico Estanqueidade e resistência à temperatura e à pressão interna Firmeza e estanqueidade da conexão do bico do recipiente com o equipo Resistência da alça de sustentação Resistência ao impacto Estanqueidade do lugar de inoculação Aderência do rótulo (etiqueta) Peso e dimensões Para o diafragma penetrável do tubo membrana: A empresa deverá comprovar que o diafragma, utilizado no tubo de conexão do equipo com a embalagem, apresenta estanqueidade, resistência à temperatura e pressão (pela passagem de fluxo ar 2 bar de pressão com o tubo inserido em um recipiente cheio de água por 30 segundos - nenhuma bolha deve ser produzida na 89
90 água) que permita avaliar se o sistema de fechamento não apresente fissuras e/ou rasuras que possam permitir a contaminação da solução Requisitos Químicos Deverão obedecer todos os requisitos químicos para material em questão constantes na edição mais atualizada da farmacopéia Britânica Requisitos Biológicos Impermeabilidade aos microorganismos Toxicidade Substâncias pirogênicas Métodos de ensaios dos recipientes plásticos IV - Apresentar os estudos de Estabilidade Acelerado e resultados parciais de Longa Duração de três lotes para o novo acondicionamento de acordo com a RE 01/05. Para os frascos ampola que mantiveram as mesmas especificações do material de embalagem, durante a adequação do sistema aberto para sistema fechado, serão aceitos os estudos realizados para sistema aberto de acordo com a RE 560/02, RE 398/04 e/ou a RE 01/05. Na primeira renovação de registro ocorrida a partir de 03/2009 serão exigidos os estudos de estabilidade de longa duração concluídos com o novo acondicionamento. Art 3º. O detentor do registro, ao peticionar o novo acondicionamento, deverá também peticionar o assunto n "Alteração do Processo Produtivo para adequação das Soluções Parenterais de Grande Volume ao Sistema Fechado", sem pagamento de taxa. Art 4º. É proibida a produção a partir do dia 13/03/2008 de Soluções Parenterais de Grande Volume em Sistema Aberto, no entanto será permitido a comercialização até 12/09/2008 dos lotes fabricados antes da data 13/03/2008. Art 5º. Para casos de exportação de Solução Parenteral de Grande Volume em Sistema Aberto, deverá protocolizar o assunto: "Autorização de Fabricação para fim Exclusivo de Exportação de Medicamento". Art 6º. A partir desta publicação não serão deferidas as petições de registro de Soluções Parenterais de Grande Volume em Sistema Aberto. Referências consultadas: Anexos E e M da Portaria 500/1997, Bristish Pharmacopeia Volume IV Appendix A Art. 7º. Esta Resolução entra em vigor na data de sua publicação. 90
91 RDC Nº 269, DE 22 DE SETEMBRO DE 2005 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11 inciso IV do Regulamento da ANVISA aprovado pelo Decreto 3.029, de 16 de abril de 1999, c/c do Art. 111, inciso I, alínea "b" 1º do Regimento Interno aprovado pela Portaria nº. 593, de 25 de agosto de 2000, republicada no DOU de 22 de dezembro de 2000, em reunião realizada em 29, de agosto de 2005, considerando a necessidade do constante perfeiçoamento das ações de controle sanitário na área de alimentos visando a promoção e proteção à saúde da população; considerando a necessidade de atualizar os valores de Ingestão Diária Recomendada (IDR) de Proteína, Vitaminas e Minerais para indivíduos e diferentes grupos populacionais; considerando a necessidade de atualizar os valores de Ingestão Diária Recomendada (IDR) de Proteína, Vitaminas e Minerais a serem utilizados como parâmetro de ingestão de nutrientes por indivíduos e diferentes grupos populacionais; considerando as diretrizes da Política Nacional de Alimentação e Nutrição sobre o controle dos distúrbios nutricionais e doenças associadas à alimentação e nutrição; adota a seguinte Resolução de Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Aprovar o "REGULAMENTO TÉCNICO SOBRE A INGESTÃO DIÁRIA RECOMENDADA (IDR) DE PROTEÍNA, VITAMINAS E MINERAIS", constante do Anexo desta Resolução. Art. 2º As empresas têm o prazo de 01 (um) ano após a data da publicação para adequarem seus produtos. Art. 3º Para os medicamentos específicos, cujos teores de vitaminas e ou minerais estiverem acima dos valores de IDR estabelecidos por esta Resolução, devem ser notificadas as alterações de bula e rótulo do medicamento à área competente desta Agência no prazo de 01 (um) ano. 1º Os medicamentos específicos categorizados neste artigo, cuja validade de registro expirar a partir de 31 de dezembro de 2006, deverão atender a Resolução ANVISA/MS RDC no 132/03. 2º As demais adequações para medicamentos específicos serão estabelecidas pela área competente desta Agência. Art. 4º O descumprimento aos termos desta Resolução constitui infração sanitária sujeitando os infratores às penalidades previstas na Lei nº , de 20 de agosto de 1977 e demais disposições aplicáveis. Art. 5º Revogam-se as disposições em contrário, em especial a Portaria SVS/MS nº 33/98. Art. 6º Esta Resolução entra em vigor na data de sua publicação. DIRCEU RAPOSO DE MELLO ANEXO REGULAMENTO TÉCNICO SOBRE INGESTÃO DIÁRIA RECOMENDADA (IDR) PARA PROTEÍNA, VITAMINAS E MINERAIS 1. Alcance Adotar os valores constantes das tabelas deste Regulamento como níveis de Ingestão Diária Recomendada (IDR) para Proteína, Vitaminas e Minerais: 91
92 Tabela 1 - Ingestão Diária Recomendada para Adultos Tabela 2 - Ingestão Diária Recomendada para Lactentes e Crianças Tabela 3 - Ingestão Diária Recomendada para Gestantes e Lactantes 2. Definição Ingestão Diária Recomendada (IDR) é a quantidade de proteína, vitaminas e minerais que deve ser consumida diariamente para atender às necessidades nutricionais da maior parte dos indivíduos e grupos de pessoas de uma população sadia. 3. REFERÊNCIAS 3.1. BRASIL. Resolução ANVISA/MS RDC nº 360, de 23 de dezembro de Regulamento Técnico sobre Rotulagem Nutricional de Alimentos Embalados. Diário Oficial da União, Brasília, DF, 26 dez Seção FAO/OMS. Human Vitamin and Mineral Requirements. In: Report 7th Joint FAO/OMS Expert Consultation. Bangkok, Thailand, xxii + 286p INSTITUTE OF MEDICINE. Food and Nutrition Board. Dietary Reference Intakes. National Academic Press, Washington D.C., INGESTÃO DIÁRIA RECOMENDADA DE PROTEÍNA, VITAMINAS E MINERAIS 4.1. Ingestão Diária Recomendada para Adultos (Tabela 1) Tabela 1 - Ingestão Diária Recomendada para Adultos Nutriente Unidade Valor Proteína (1) g 50 Vitamina A (2) (a) micrograma RE 600 Vitamina D (2) (b) micrograma 5 Vitamina C (2) mg 45 Vitamina E (2) (c) mg 10 Tiamina (2) mg 1,2 Riboflavina (2) mg 1,3 Niacina (2) mg 16 Vitamina B6 (2) mg 1,3 Ácido fólico (2) micrograma 240 Vitamina B12 (2) micrograma 2,4 Biotina (2) micrograma 30 92
93 Ácido pantotênico (2) mg 5 Vitamina K (2) micrograma 65 Colina (1) mg 550 Cálcio (2) mg 1000 Ferro (2) (d) mg 14 Magnésio (2) mg 260 Zinco (2) (e) mg 7 Iodo (2) micrograma 130 Fósforo (1) mg 700 Flúor (1) mg 4 Cobre (1) micrograma 900 Selênio (2) micrograma 34 Molibdênio (1) micrograma 45 Cromo (1) micrograma 35 (a) 1 micrograma retinol = 1 micrograma RE; 1 micrograma beta-caroteno = 0,167 micrograma RE; 1 micrograma de outros carotenóides provitamina A = 0,084 micrograma RE; 1 UI = 0,3 micrograma de retinol equivalente (2). (b) 1 micrograma de colicalciferol = 40 UI. (c) mg alfa-te/dia; 1,49 UI = 1mg d-alfa-tocoferol (1). (d) 10% de Biodisponibilidade (e) Biodisponibilidade moderada - calculada com base em dietas mistas contendo proteína de origem animal INSTITUTE OF MEDICINE. Food and Nutrition Board. Dietary Reference Intakes. National Academic Press, Washington D.C., FAO/OMS. Human Vitamin and Mineral Requirements. In: Report 7th Joint FAO/OMS Expert Consultation. Bangkok, Thailand, xxii + 286p Ingestão Diária Recomendada para Lactentes e Crianças (Tabela 2) Tabela 2 - Ingestão Diária Recomendada para Lactentes e Crianças Nutriente Unidade Lactente Crianças 0-6 meses 7-11 meses 1-3 anos 4-6 anos (37 meses a 6 anos) 7-10 anos Proteína (1) g 9,
94 Vitamina A (2) (a) Vitamina D (2) (b) micrograma RE micrograma Vitamina C (2) mg Vitamina E (2) c mg 2,7 2, Tiamina (2) mg 0,2 0,3 0,5 0,6 0,9 Riboflavina (2) mg 0,3 0,4 0,5 0,6 0,9 Niacina (2) mg Vitamina B6 (2) Ácido fólico (2) Vitamina B12 (2) Ácido pantotênico (2) mg 0,1 0,1 0,5 0,5 1,0 micrograma micrograma 0,4 0,5 0,9 1,2 1,8 mg 1,7 1, Vitamina K (2) micrograma Colina (1) mg Cálcio (2) mg Ferro (2) (d) mg 0, Magnésio (2) mg Zinco (2)(e) mg 2,8 4,1 4,1 5,1 5,6 Iodo (2) micrograma Fósforo (1) mg Flúor (1) mg 0,01 0,5 0,7 1 2 Cobre (1) micrograma Selênio (2) micrograma Molibdênio (1) micrograma
95 Cromo (1) micrograma 0,2 5, Manganês (1) mg 0,003 0,6 1,2 1,5 1,5 (a) 1 micrograma retinol = 1 micrograma RE; 1 micrograma beta-caroteno = 0,167 micrograma RE; 1 micrograma de outros carotenóides provitamina A = 0,084 micrograma RE; 1 UI = 0,3 micrograma de retinol equivalente (2). (b) 1 micrograma de colicalciferol = 40 UI. (c) mg alfa-te; 1,49 UI = 1mg d-alfa-tocoferol (1). (d) 10% de Biodisponibilidade (e) Biodisponibilidade moderada - calculada com base em dietas mistas contendo proteína de origem animal (1) INSTITUTE OF MEDICINE. Food and Nutrition Board. Dietary Reference Intakes. National Academic Press, Washington D.C., (2) FAO/OMS. Human Vitamin and Mineral Requirements. In: Report 7th Joint FAO/OMS Expert Consultation. Bangkok, Thailand, xxii + 286p Ingestão Diária Recomendada para Gestantes e Lactantes (Tabela 3) Tabela 3 - Ingestão Diária Recomendada para Gestantes e Lactantes Nutriente Unidade Gestante Lactante Proteína (1) g Vitamina A (2) (a) Vitamina D (2) (b) micrograma RE micrograma 5 5 Vitamina C (2) mg Vitamina E (2) c mg Tiamina (2) mg 1,4 1,5 Riboflavina (2) mg 1,4 1,6 Niacina (2) mg Vitamina B6 (2) Ácido fólico (2) Vitamina B12 (2) mg 1,9 2,0 micrograma micrograma 2,6 2,8 95
96 Ácido pantotênico (2) mg 6 7 Vitamina K (2) micrograma Colina (1) mg Cálcio (2) mg Ferro (2) (d) mg Magnésio (2) mg Zinco (2)(e) mg 11 9,5 Iodo (2) micrograma Fósforo (1) mg Flúor (1) mg 3 3 Cobre (1) micrograma Selênio (2) micrograma Molibdênio (1) micrograma Cromo (1) micrograma Manganês (1) mg 2,0 2,6 (a) 1 micrograma retinol = 1 micrograma RE; 1 micrograma beta-caroteno = 0,167 micrograma RE; 1 micrograma de outros carotenóides provitamina A = 0,084 micrograma RE; 1 UI = 0,3 micrograma de retinol equivalente (2). (b) 1 micrograma de colicalciferol = 40 UI. (c) mg alfa-te; 1,49 UI = 1mg d-alfa-tocoferol (1). (d) 10% de Biodisponibilidade (e) Biodisponibilidade moderada - calculada com base em dietas mistas contendo proteína de origem animal (1) INSTITUTE OF MEDICINE. Food and Nutrition Board. Dietary Reference Intakes. National Academic Press, Washington D.C., (2) FAO/OMS. Human Vitamin and Mineral Requirements. In: Report 7th Joint FAO/OMS Expert Consultation. Bangkok, Thailand, xxii + 286p. 96
97 RDC nº 8, de 2 de janeiro de 2001 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11, inciso IV, do Regulamento da ANVISA aprovado pelo Decreto 3.029, de 16 de abril de 1999, em reunião realizada em 28 de dezembro de 2000, Adotou a seguinte Resolução de Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Aprovar o Regulamento Técnico que Institui as Boas Práticas de Fabricação do Concentrado Polieletrolíticos para Hemodiálise - CPHD. Art. 2º Fica concedido prazo de 180 (CENTO E OITENTA) dias para o cumprimento do Regulamento Técnico, objeto desta Resolução. 1º Durante o prazo a que se refere o artigo anterior os estabelecimentos em funcionamento deverão ser avaliados pelas autoridades sanitárias locais. 2º Findo o prazo estabelecido no caput deste artigo os estabelecimentos infratores ficarão sujeitos às penalidades previstas na Lei nº 6437, de 20 de agosto de Art. 3º Os processos, de registro de CPHD, que já se encontram tramitando na Agência Nacional de Vigilância Sanitária devem atender às exigências previstas nesta Resolução, a partir de sua publicação. Art. 4º Esta Resolução de Diretoria Colegiada entra em vigor na data de sua publicação. GONZALO VECINA NETO ANEXO - REGULAMENTO TÉCNICO QUE INSTITUI AS BOAS PRÁTICAS DE FABRICAÇÃO DO CONCENTRADO POLIELETROLÍTICOS PARA HEMODIÁLISE - CPHD. 1. OBJETIVO: Este Regulamento Técnico fixa os requisitos mínimos exigidos para as Boas Práticas de Fabricação do Concentrado Polieletrolítico para Hemodiálise - CPHD. 2. REFERÊNCIAS: 2.1. ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS-ABNT (Brasil). Emprego de cores para identificação de tubulação NBR 6493:, Rio de Janeiro [1994]. 5 p ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS-ABNT (Brasil). Sinalização de Segurança - NB 26, Rio de Janeiro p BOAS Práticas para Fabricação de Produtos Farmacêuticos. Brasília: Ministério da Saúde, p BRASIL. Lei nº 8.078, de 11 de setembro de Código de Defesa do Consumidor. Diário Oficial [da República Federativa do Brasil], Brasília, v.128, n. 176, supl., p. 1 12, 12 set BRASIL. Portaria nº 3.214, de 08 de junho de Sinalização de Segurança NR 26. Diário Oficial [da República Federativa do Brasil], Brasília, V. 116, n. 127, p , 06 jul BRASIL. Portaria nº 8, de 08 de maio de Altera norma regulamentadora NR 07. Programa de Controle Médico de Saúde Ocupacional. Diário Oficial [da República Federativa do Brasil], Brasília, V. 134, n 91, p. 8202, de 13 de maio de BRASIL. Portaria nº 1884, de 11 de novembro de Aprova normas destinadas ao planejamento, exame e aprovação de Projetos Físicos de Estabelecimentos Assistenciais de Saúde. Diário Oficial [da República Federativa do Brasil], Brasília, 15 dez
98 2.8. BRASIL. Portaria GM/MS nº 1179 de 17 de junho de Denominação Comum Brasileira DCB. Diário Oficial [da República Federativa do Brasil], Brasília, 18 jun BRITISH Pharmacopea, EUROPEAN Pharmacopeia, USP 24/NF 18, WATER, Purified. European Pharmacopeia p DEFINIÇÕES: Para efeito deste Regulamento Técnico são adotadas as seguintes definições; 3.1. Calibração conjunto de operações que estabelece, em condições específicas, a correspondência entre o estímulo e a resposta de um instrumento Concentrado Polieletrolítico para Hemodiálise CPHD: Concentrado de eletrólitos, com ou sem glicose, apresentado na forma sólida ou líquida para ser empregado na terapia de diálise renal, após diluição recomendada pelo fabricante e utilizando equipamento específico Controle de Qualidade: conjunto de operações (programação, coordenação e execução) com o objetivo de verificar a conformidade das matérias primas, materiais de embalagem e do produto acabado com as especificações estabelecidas Data de Validade: data impressa no recipiente ou no rótulo do produto, informando o tempo durante o qual se espera que o mesmo mantenha as especificações estabelecidas, desde que estocado nas condições recomendadas e, após o qual não deve ser usado Dialisato: é a solução obtida após diluição do CPHD, na proporção adequada para uso Embalagem Primária: recipiente que fica em contato direto com o produto Embalagem Secundária: a que protege a embalagem primária para o transporte, armazenamento e distribuição Fórmula Padrão: documento ou grupo de documentos que especificam as matérias-primas com respectivas quantidades e os materiais de embalagem, juntamente com a descrição dos procedimentos, incluindo instruções sobre o controle em processo, e precauções necessárias para a produção de determinada quantidade (lote) de um produto Lote ou partida: quantidade de um produto, obtido em uma única preparação, cuja característica essencial é a homogeneidade Material de Embalagem: Recipientes, rótulos e caixas para acondicionamento de CPHD Número de Lote: designação impressa em cada unidade do recipiente constituída de combinações de letras, números ou símbolos Ordem de Produção: documento administrativo que estabelece a programação sequencial de produção Prazo de Validade: período de tempo (em dias, meses ou anos) durante o qual o produto se mantém dentro dos limites especificados de pureza, qualidade e identidade, na embalagem adotada e se estocado nas condições recomendadas no rótulo Rótulo: identificação impressa ou litogravada, bem como os dizeres pintados ou gravados a fogo, pressão ou decalco, aplicado diretamente sobre a embalagem primária e secundária do produto Unidade Formadora de Colônia - UFC: colônias isoladas de microrganismos viáveis, passíveis de contagem e obtidas a partir da semeadura, em meio de cultura específico Validação Programa documentado para a comprovação e verificação da efetividade e reprodutibilidade de um processo, operação e método analítico. 4. ABRANGÊNCIA 98
99 Exclui-se do cumprimento do ANEXO II (DIRETRIZES PARA REGISTRO DE CONCENTRADOS POLIELETROLÍTICOS PARA HEMODIÁLISE CPHD) deste Regulamento o CPHD produzido em farmácias hospitalares e de clínicas especializadas, exclusivamente para uso próprio, mas devem atender todos os princípios das Boas Práticas de Fabricação deste Regulamento, no que couber. 5. CONDIÇÕES GERAIS As Boas Práticas de Fabricação do Concentrado Polieletrolítico para Hemodiálise - BPFCPHD estabelecem as orientações gerais para aplicação nas operações de fabricação, aquisição de matérias primas e materiais de embalagem. É indispensável à efetiva inspeção durante todo o processo de fabricação de CPHD, de modo a garantir a qualidade do produto ORGANIZAÇÃO E PESSOAL Estrutura Organizacional Todo fabricante de CPHD deve ter um organograma que demonstre possuir estrutura organizacional e de pessoal suficiente para garantir que os produtos, por ele fabricados, estejam de acordo com os requisitos deste Regulamento Todo fabricante de CPHD deve contar com pessoal qualificado e em quantidade suficiente para o desempenho de todas as tarefas preestabelecidas, para que todas as operações sejam executadas corretamente Responsabilidades O responsável técnico pela fabricação de CPHD é o farmacêutico Os responsáveis pelo controle da qualidade e pela produção devem possuir conhecimentos científicos e experiência prática na atividade As atribuições e responsabilidades individuais devem estar formalmente descritas e perfeitamente compreendidas pelos envolvidos Na aplicação das Boas Práticas de Fabricação do Concentrado Polieletrolíticos para Hemodiálise - CPHD é recomendável não haver sobreposição nas responsabilidades do pessoal Treinamento Deve haver um programa de treinamento, com os respectivos registros, para todo o pessoal envolvido nas atividades que podem afetar a qualidade da fabricação de CPHD Os funcionários devem receber treinamento inicial e contínuo, inclusive instruções de higiene relevantes às suas atividades, além de motivação para a manutenção dos padrões de qualidade Todo pessoal deve conhecer os princípios das Boas Práticas de Fabricação do Concentrado Polieletrolítico para Hemodiálise BPFCPHD Saúde, Higiene e Conduta A admissão dos funcionários deve ser precedida de exames médicos, sendo obrigatória a realização de avaliações médicas periódicas dos funcionários diretamente envolvidos na fabricação de CPHD, atendendo à NR n.º 7 - MT - Programa de Controle Médico de Saúde Ocupacional - PCMSO O pessoal em contato com o produto ou com o ambiente de fabricação deve apresentar boas condições de saúde e higiene Os portadores de enfermidades infectocontagiosas devem ser afastados temporariamente ou definitivamente das suas atividades Todos os funcionários devem ser orientados quanto às práticas de higiene pessoal e lavagem das mãos Ao pessoal envolvido na fabricação de CPHD, particularmente na área de preparação/envasamento, não deve ser permitido o uso de cosméticos e objetos de adorno de uso pessoal. 99
100 Os procedimentos de higiene pessoal e a utilização de roupas protetoras devem ser exigidos a todas as pessoas para entrarem na área de preparação/envasamento, sejam elas funcionários, visitantes, administradores e inspetores Vestuário A paramentação para a entrada na área de produção deve ser realizada em área especificamente designada para vestiário O pessoal envolvido na fabricação e controle da qualidade de CPHD tem que dispor de uniformes que devem ser trocados para garantir a higiene apropriada O pessoal de embalagem e transporte interno, assim como os auxiliares indiretos (manutenção), devem usar uniformes igualmente limpos Infraestrutura física O fabricante de CPHD deve atender aos requisitos quanto à infraestrutura física deste Regulamento Técnico, e estar em conformidade com os critérios de circulações internas e externas, de instalações prediais e de condições ambientais de conforto e de segurança Ambientes Uma unidade destinada à fabricação de CPHD deve possuir, no mínimo, os seguintes ambientes: a) Área de armazenamento para matéria prima e material de embalagem; b) Sala de pesagem de matéria prima; c) Vestiário; d) Sala de preparação/envasamento de CPHD; e) Sala de rotulagem/embalagem; f) Área de armazenamento de produto acabado/expedição; g) Área de Controle de Qualidade h) Sanitários de funcionários (masculino e feminino); i) DML - depósito de material de limpeza Para a manipulação em farmácias privativas de hospitais ou clínicas que produzem CPHD as salas especificadas acima podem ser consideradas como áreas, desde que bem demarcadas e a movimentação de materiais possa ser feita de modo seguro, atendendo aos requisitos deste regulamento Características Gerais Os ambientes destinados à fabricação de CPHD devem se adequar às operações desenvolvidas e assegurar a qualidade do produto Os ambientes destinados à fabricação de CPHD devem possuir superfícies internas (pisos, paredes e teto) lisas e impermeáveis, sem rachaduras, resistentes aos agentes sanitizantes e facilmente laváveis Os ambientes devem ter dimensões suficientes ao desenvolvimento das operações, dispondo de todos os equipamentos e materiais de forma organizada e racional, objetivando evitar os riscos de contaminação, misturas de componentes e garantir a sequência das operações Todos os ralos de esgotos devem ser sifonados e com tampas escamoteáveis Os ambientes devem ser protegidos contra a entrada de aves, insetos, roedores e poeira A iluminação e a ventilação devem ser suficientes e adequadas As instalações de água potável devem ser construídas de materiais impermeáveis, para evitar infiltração e facilitar a limpeza e inspeções periódicas Os reservatórios de água potável devem ser devidamente protegidos para evitar contaminações por microrganismos, insetos ou aves. 100
101 Os vestiários, lavatórios e os sanitários devem ser de fácil acesso e suficientes para o número de funcionários Os sanitários não devem ter comunicação direta com a sala preparação/envasamento e armazenamento Salas de descanso e refeitório, quando existirem, devem ser separadas das demais áreas Condições Específicas Áreas de Armazenamento As áreas de armazenamento são restritas às pessoas autorizadas e devem ter capacidade suficiente para assegurar a estocagem ordenada das matérias primas, materiais de embalagem e CPHD pronta Os materiais devem ser estocados em locais identificados, de modo a facilitar a sua localização para uso, sem riscos de troca Deve existir local para segregar matérias primas, materiais de embalagem e CPHD reprovados, recolhidos ou devolvidos Deve existir área demarcada ou um sistema validado que garanta a quarentena de matérias primas, materiais de embalagem e CPHD Todos os materiais devem ser armazenados sob condições apropriadas, de modo a preservar a identidade e integridade dos mesmos, e de forma ordenada, para que possa ser feita a separação dos lotes e a rotação do estoque, obedecendo à regra: primeiro que entra, primeiro que sai As condições de temperatura para armazenamento de CPHD, pronto para transporte, devem atender às recomendações estabelecidas pelo fabricante e expressa no rótulo do produto, de modo a garantir a estabilidade do mesmo Os materiais de limpeza e germicidas em estoques devem ser armazenados em área especificamente designada e identificada Vestiário Sala destinada à paramentação tendo, preferencialmente, dois ambientes e ser ventilada O vestiário deve possuir lavatório, com provisão de sabão líquido e, opcionalmente um antisséptico, além de recursos para secagem das mãos O vestiário deve funcionar como acesso para as salas de pesagem e preparação/envasamento Sala de Pesagem de Matérias Primas A sala destinada à pesagem de matérias primas deve possuir uma ante sala destinada à limpeza prévia das embalagens para entrada na sala de pesagem A sala de pesagem deve ser contígua à sala de preparação/envasamento e dotada de passagem para a entrada da matéria prima em condições de segurança A sala de pesagem deve ter dimensões e instalações compatíveis com o volume de matérias primas Sala de Preparação/Envasamento Sala independente e destinada, exclusivamente, para este fim, livre de trânsito de materiais e pessoas estranhas ao setor A sala de preparação/envasamento deve ser mantida em rigorosas condições de limpeza e higiene A sala deve dispor de ponto de água purificada para lavagem de equipamentos, preparação de CPHD e lavagem dos recipientes para o envasamento O mobiliário deve ser o mínimo e estritamente necessário ao trabalho aí desenvolvido e ser construído de material liso, impermeável e facilmente lavável Depósito de Material de Limpeza - DML Sala destinada exclusivamente à guarda de material de limpeza e sanitização em uso nos ambientes. Deve possuir tanque para a lavagem do material de limpeza. 101
102 5.3. EQUIPAMENTOS E MOBILIÁRIOS Localização e instalação dos equipamentos Os equipamentos devem ser localizados, instalados e mantidos de forma a estarem adequados às operações a serem realizadas A estrutura dos equipamentos deve visar à minimização dos riscos de erro e permitir que os mesmos sejam efetivamente limpos e assim mantidos para que seja evitada a contaminação cruzada Os equipamentos utilizados na fabricação de CPHD devem estar instalados de forma que, periodicamente, possam ser fácil e totalmente limpos As tubulações devem ser claramente identificadas, conforme norma específica Os instrumentos e os equipamentos do laboratório de controle de qualidade devem ser compatíveis com os procedimentos analíticos inerentes ao produto fabricado Calibração e Verificação dos Equipamentos As balanças empregadas na pesagem das matérias primas e equipamentos do controle de qualidade, quando aplicável, devem ser calibrados e periodicamente verificados, conforme procedimentos escritos A calibração deve ser executada por pessoal capacitado, utilizando padrões rastreáveis à Rede Brasileira de Calibração, com procedimentos reconhecidos oficialmente, no mínimo uma vez ao ano Em função da frequência de uso dos equipamentos e dos registros das verificações dos mesmos, deve ser estabelecida a periodicidade da calibração A verificação dos equipamentos deve ser feita por pessoal treinado, empregando procedimentos escritos, com orientação específica e limites de tolerância definidos Deve haver registros das calibrações e verificações realizadas As etiquetas com datas referentes à última e à próxima calibração devem estar afixadas no equipamento Manutenção Todos os equipamentos devem ser submetidos à manutenção preventiva, de acordo com um programa formal, e corretiva, quando necessário, obedecendo a procedimentos operacionais escritos, com base nas especificações dos manuais dos fabricantes Devem existir registros das manutenções preventivas e corretivas realizadas Limpeza e Sanitização Programas e procedimentos operacionais de limpeza e sanitização das áreas, instalações, equipamentos e materiais devem estar disponíveis ao pessoal responsável e operacional Os produtos usados na limpeza e sanitização não devem contaminar, com substâncias tóxicas, químicas, voláteis e corrosivas, as instalações e os equipamentos de preparação Antes e após, cada sessão de fabricação, a área de preparação/envasamento e os equipamentos devem ser limpos e sanitizados, efetuando-se os respectivos registros desses procedimentos Antes do início do trabalho de fabricação deve ser verificada a condição de limpeza dos equipamentos e os respectivos registros MATERIAIS Para efeito deste Regulamento, inclui-se no item materiais: matérias primas, materiais de embalagem e saneantes empregados no processo de fabricação de CPHD Aquisição Compete ao farmacêutico o estabelecimento de critérios e a supervisão do processo de aquisição Deve haver especificação técnica detalhada de todos os materiais necessários à fabricação de CPHD, de modo a garantir que a aquisição atenda corretamente aos padrões de qualidade estabelecidos. 102
103 As matérias primas devem ser especificadas quanto a sua pureza física e química, teor do princípio ativo e pureza microbiológica que garanta no máximo 100 UFC por ml ou g no produto final Os materiais devem ser adquiridos somente de fornecedores qualificados quanto aos critérios de qualidade A qualificação do fornecedor de materiais deve abranger, no mínimo, os seguintes critérios: a) exato atendimento às especificações estabelecidas com base nas monografias farmacopeias. b) os materiais devem ter registro ou serem declarados isentos de registro pela Agência Nacional de Vigilância Sanitária. c) efetivo envio de certificado de análise dos lotes fornecidos, comprovando o atendimento às especificações estabelecidas. d) avaliação do histórico de fornecimento A qualificação de fornecedores deve ser documentada quanto ao procedimento utilizado, com os respectivos registros A quantidade adquirida dos materiais deve levar em consideração o consumo médio, o prazo de validade dos mesmos e a capacidade da área de estocagem nas condições exigidas Os recipientes adquiridos e destinados ao envasamento de CPHD devem ser atóxicos, compatíveis físico - quimicamente com a composição do seu conteúdo e devem manter a qualidade e estabilidade das CPHD durante o seu armazenamento e transporte A água é considerada como matéria prima e deve atender as especificações para água purificada, conforme item Recebimento (inspeção, aprovação, reprovação) O recebimento dos materiais deve ser realizado por pessoa treinada, com conhecimentos específicos sobre os mesmos, segundo os requisitos previamente estabelecidos Todos os materiais devem ser submetidos à inspeção de recebimento, devidamente documentada, para verificar a integridade e condições de limpeza da embalagem e quanto à correspondência entre o pedido, a nota de entrega e os rótulos do material recebido Qualquer problema constatado na inspeção do recebimento que possa afetar a qualidade do produto deve ser analisada pelo farmacêutico para orientar as providências a serem tomadas Se uma única remessa de material contiver lotes distintos, cada lote deve ser levado em consideração separadamente para inspeção e liberação CONTROLE DO PROCESSO DE FABRICAÇÃO Preparação (material, pessoal, processo e inspeção) A preparação de CPHD deve atender a uma ordem de fabricação por lote a ser produzido e as operações do processo devem seguir as orientações de Ficha Técnica de Fabricação, informatizada ou não, onde devem ser registrados todos os dados importantes, que possibilitem o rastreamento da fabricação Devem existir procedimentos operacionais escritos para todas as etapas do processo de preparação Todas as embalagens de matérias primas e recipientes devem ser limpos antes da entrada na área de preparação Deve ser efetuado o registro do número do lote de cada uma das matérias primas utilizadas na preparação de CPHD, de forma a possibilitar o rastreamento O transporte dos materiais limpos para a sala de preparação/envasamento deve ser efetuado de modo a não afetar a qualidade do produto Todas as superfícies de trabalho devem ser limpas e sanitizadas, antes e depois de cada sessão de preparação Devem existir registros das operações de limpeza dos equipamentos empregados no processo de fabricação de CPHD. 103
104 Todo pessoal envolvido no processo de preparação de CPHD deve proceder à lavagem das mãos e antebraços, antes do início de qualquer atividade na área de preparação O envasamento de CPHD deve ser feito em recipiente que atenda os requisitos para uso parenteral, conforme especificado na farmacopeia europeia, 1997 e suas atualizações, e garanta a estabilidade físico-química do produto, não devendo ser reutilizado A quantidade envasada não deve ser menor que 100% e não maior que 104 % do peso ou volume declarado Rotulagem e Embalagem Devem existir procedimentos operacionais escritos para as operações de rotulagem e embalagem de CPHD , O rótulo deve apresentar as seguintes características: a) Em todos os concentrados o rótulo deve apresentar a tarja vermelha (Pantone Vermelho 485 C), com largura correspondente a um quinto da maior face do rótulo, com os dizeres "uso sob prescrição médica". Deverá ser respeitado o limite mínimo de 10 mm nas bases das rotulagens, como caracterização daquilo que se entende como rodapé, após o qual deverá ser adotada a faixa vermelha dos medicamentos de uso sob prescrição médica. b) Instituir, uma tarja no cabeçalho, com largura correspondente a um quinto da menor face do rótulo, de cor azul (pantone blue 072 c) nos concentrados básicos. c) Nome Comercial: c.1) Pode ser adotado o nome do produto Concentrado Polieletrolítico para Hemodiálise ou CPHD, precedido do nome ou logomarca da empresa, ou; c.2.) Nome de marca definido para o produto, se houver; d) Nome do Produto: Concentrado Polieletrolítico para Hemodiálise ou CPHD; e) Nome Genérico: é o nome dos dois primeiros sais da formulação seguido da expressão: "+ Associações"; f) Volume ou peso declarado; g) Fórmula completa com os componentes, de acordo com a DCB ou CAS (Tabela I), e as quantidades dos sais expressas em p/v (g/l) ou p/p (g/g) no concentrado e meq/l dos íons ou mmol/l das moléculas, após diluição, atendendo aos limites estabelecidos na Tabela II; h) Instruções de uso, incluindo a proporção de diluição a ser empregada. i) Restrições, precauções e cuidados que devem ser considerados; j) Destacar, em negrito, no Rótulo do produto os dizeres USO RESTRITO EM HEMODIÁLISE; k) Número de Lote, data de fabricação, prazo de validade e cuidados de conservação; l) Os dizeres legais necessários (nome da empresa, CGC, endereço, registro no Ministério da Saúde, nome do farmacêutico responsável com CRF e outros); m) Pode-se complementar qualquer informação, quando necessário, acrescentando uma bula; A embalagem primária de CPHD sob a forma líquida deve possuir tampa com lacre de segurança e a forma sólida deve ser hermeticamente selada, não devendo ser reutilizado O CPHD já rotulado deve ser acondicionado em embalagem secundária para manter a integridade do produto durante o transporte Armazenamento e Transporte As condições de armazenamento e transporte devem ser estabelecidas pelo fabricante de modo a assegurar as características do CPHD até sua utilização, desde que mantidas na faixa de 10 C a 35 C O farmacêutico é responsável pela manutenção da qualidade do CPHD até a sua entrega ao usuário O CPHD não deve ser armazenado ou transportado com os seguintes produtos: a) alimentos e materiais perecíveis; b) animais; c) solventes orgânicos; 104
105 d) gases; e) substâncias corrosivas ou tóxicas; f) pesticidas e agrotóxicos; g) materiais radioativos No armazenamento e transporte deve ser respeitado o empilhamento máximo recomendado pelo fabricante GARANTIA DA QUALIDADE Considerações Gerais A Garantia da Qualidade tem como objetivo assegurar que os produtos e serviços estejam dentro dos padrões de qualidade exigidos Para atingir os objetivos da Garantia da Qualidade na fabricação de CPHD, o fabricante deve possuir um Sistema de Garantia da Qualidade (SGQ), monitorado através de auditorias da Qualidade, e que incorpore as diretrizes das BPFCPHD com efetivo controle de qualidade, totalmente documentado Um Sistema de Garantia da Qualidade apropriado para a fabricação de CPHD deve assegurar que: a) as operações de fabricação de CPHD sejam claramente especificadas por escrito e que as exigências de BPFCPHD sejam cumpridas. b) os controles de qualidade necessários para avaliar as matérias primas, o processo de fabricação, conservação e transporte de CPHD, sejam realizados de acordo com procedimentos escritos e devidamente registrados. c) os equipamentos e instrumentos sejam calibrados, com documentação comprobatória. d) o CPHD só deve ser comercializado após o farmacêutico responsável ter atestado formalmente, por meio de um documento de liberação, que o produto foi fabricado dentro dos padrões especificados pelas BPFCPHD. e) o CPHD seja armazenado e transportado de forma que a qualidade do mesmo seja mantido até a sua entrega ao usuário, proibida a reutilização da embalagem. f) sejam realizadas auditorias da qualidade para avaliar regularmente o Sistema de Garantia da Qualidade e oferecer subsídios para a implementação de ações corretivas, de modo a assegurar um processo de melhoria contínua. g) seja proibida a reutilização da embalagem primária Controle de Qualidade do CPHD O Controle de Qualidade deve avaliar todos os aspectos relativos às matérias primas, materiais de embalagem, procedimentos de limpeza, higiene e sanitização, armazenamento e transporte do CPHD, de modo a garantir que suas especificações e critérios estabelecidos por este Regulamento estejam atendidos As matérias primas e materiais de embalagem devem ser inspecionados no recebimento para verificar a integridade física da embalagem e as informações dos rótulos O certificado de análise de cada matéria prima emitido pelo fabricante deve ser avaliado para verificar o atendimento às especificações estabelecidas As matérias primas devem atender às monografias da Farmacopeia Brasileira ou outro compêndio oficial reconhecido pela Agência Nacional de Vigilância Sanitária e serem avaliadas quanto a pureza microbiológica e endotoxinas bacterianas Desde que o fornecedor esteja devidamente qualificado para o fornecimento de determinada matéria prima, além da inspeção do recebimento, devidamente documentada e registrada, cada lote recebido deve ser, no mínimo, avaliado quanto à identidade, pureza microbiológica e endotoxinas bacterianas, A água para fabricação de CPHD deve ser obtida através do tratamento da água potável, em sistema (por destilação, troca-iônica ou osmose reversa) validado que garanta a obtenção de água purificada, atendendo as especificações da Tabela III O sistema de tratamento da água deve ser especificado e dimensionado e com a sua qualidade garantida através de validação dos parâmetros microbiológicos e físico-químicos. 105
106 A verificação da pureza da água é de responsabilidade das empresas, que devem emitir laudos próprios adequados ou de serviços de terceirizados, atendendo aos limites e periodicidade de análises estabelecidos neste Regulamento (Tabela III). TABELA III Componentes Limites Máximos Periodicidade Aspecto Límpido, incolor, inodoro. Diário * ph 5 10 Diário * Condutividade a 25 C 3,0 ms/cm-1. Diário * Cloreto 50,0 mg/l (ppm) Diário * Sulfato 50,0 mg/l (ppm) Mensal * Amônia 0,2 mg/l (ppm) Mensal * Cálcio 2,0 mg/l (ppm) Mensal * Cloro Residual 0,1 mg/l (ppm) Semanal * Alumínio 0,01 mg/l (ppm) Mensal * Magnésio 2,0 mg/l (ppm) Mensal * Mercúrio 0,001 mg/l (ppm) Mensal * Potássio 2,0 mg/l (ppm) Semanal * Sódio 50,0 mg/l (ppm) Semanal * Zinco 0,1 mg/l (ppm) Mensal 106
107 * Nitrato 2,0 mg/l (ppm) Diário * Metais Pesados 0,1 mg/l (ppm) em Pb Mensal * Substâncias Oxidáveis 0,3 mg/l (ppm) Mensal Sólidos Totais 10,0 mg/l (ppm) Mensal * Contagem microbiana 100 UFC / ml Diário * Coliformes totais Ausentes Diário * Endotoxinas 0,25 U.I. / ml Diário NOTA: 1 ng de endotoxina bacteriana equivale a 5 U.I. (Unidades Internacionais) Os valores máximos correspondem à interpretação quantitativa dos padrões para Água Purificada, pela Farmacopeia Europeia, A determinação de Cloretos, Sulfatos, Amônia, Sódio, Cálcio e Dióxido de Carbono podem ser substituídos pela determinação da condutividade em três estágios, conforme USP XXIV A determinação de substâncias oxidáveis pode ser substituída pela de TOC Carbono orgânico total, conforme USP XXIV A validação do sistema de tratamento de água deve ser realizada uma vez por ano ou sempre que houver qualquer alteração que possa interferir na qualidade da água Os procedimentos de limpeza, higiene e sanitização devem ser desenvolvidos e monitorados para verificar o cumprimento dos requisitos estabelecidos O CPHD deve ser submetido aos seguintes controles: a) inspeção visual em 100% das amostras, para assegurar a integridade física da embalagem; b) verificação da exatidão das informações do rótulo; c) aspecto: o CPHD líquido deve ser límpido, de incolor a levemente amarelado, com odor caraterístico e ausência de partículas visíveis a olho nu. O CPHD sólido deve ser um particulado ou granulado uniforme, sem elementos estranhos, de cor e odor característicos e facilmente solúvel em água; d) A quantidade contida nos recipientes não deve ser inferior à quantidade declarada, sendo aceitável uma variação positiva de até 4% (100 a 104 %) O CPHD, diluído com a água purificada empregada na preparação (Tabela III), na proporção indicada para uso, deve ser submetido aos seguintes controles: a) condutividade entre 11 ms.cm-1.e 15 ms.cm-1; b) ph entre 6,8 e 7,5; c) contagem microbiana 100 UFC/ml; d) endotoxinas bacterianas 0,5 UE / ml, até o término do prazo de validade citado pelo fabricante. 107
108 e) teores dos componentes entre + 2,5 % para o sódio e + 5% para os demais sais em relação ao declarado no rótulo do produto De cada lote de CPHD produzido deve ser separado uma alíquota mínima de 50 ml retirada da embalagem original, devidamente lacrada e identificada com o rótulo correspondente ao lote, para arquivo referência, sendo mantida por até seis meses após o prazo de validade As condições de conservação e transporte devem ser verificadas periodicamente para assegurar a manutenção das características do CPHD Todas as avaliações exigidas devem ser devidamente registradas, permitindo o seu rastreamento Prazo de validade Todo CPHD deve apresentar no rótulo um prazo de validade com indicação das condições para seu armazenamento A determinação do prazo de validade pode ser baseada em informações de avaliações da estabilidade físicoquímica das matérias primas, e ou através de realização de testes de estabilidade Fontes de informações sobre a estabilidade físico-química das matérias primas deve incluir: referências de compêndios oficiais, recomendações dos fabricantes dos mesmos e pesquisas publicadas Na interpretação das informações sobre a estabilidade das matérias primas devem ser considerados todos os aspectos de acondicionamento e conservação Os estudos de estabilidade devem ser realizados de acordo com uma programação escrita que abranja: a) Descrição da composição do CPHD b) Indicação de todos os parâmetros e métodos de teste que evidenciem a estabilidade do CPHD quanto às suas características físicas, químicas e microbiológica. c) Indicação do tempo e das condições especiais de armazenamento, transporte, abrangidos pelo estudo. d) Os testes devem ser realizados considerando as condições da Zona 4 estabelecida pela USP-XXIV, 30º C e 70% umidade relativa. e) Registro de todos os dados obtidos, com avaliação e conclusão dos estudos Os estudos de estabilidade acelerada ou de prateleira (natural) devem ser realizados em três lotes distintos de CPHD Ocorrendo mudança significativa no procedimento de fabricação ou nas matérias primas e material de embalagem que possa afetar a estabilidade do CPHD e, portanto, alterar o seu prazo de validade, deve ser realizado novo estudo de estabilidade Reclamações Toda reclamação referente ao desvio de qualidade do CPHD deve ser feita por escrito, registrada e analisada pelo fabricante A reclamação de qualidade do CPHD deve incluir nome e dados da unidade hospitalar ou do médico, nome do produto, número do lote, natureza da reclamação e responsável pela reclamação O fabricante, ao analisar a reclamação deve estabelecer as investigações a serem efetuadas e os responsáveis pelas mesmas As investigações e suas conclusões, bem como as ações corretivas implantadas, devem ser registradas O fabricante, com base nas conclusões da investigação, deve prestar esclarecimentos por escrito ao reclamante Em caso de não ser necessária a investigação, o registro deve incluir a razão pela qual a investigação foi considerada desnecessária Quando for identificada situação de risco à saúde, o fabricante fica obrigado a notificar imediatamente o órgão de Vigilância Sanitária competente. 108
109 Documentação A documentação tem como objetivo definir as especificações de todas as matérias primas e materiais de embalagem, os métodos de preparação e controle de CPHD, para a perfeita orientação do pessoal envolvido nas operações A documentação deve garantir a disponibilidade de todas as informações necessárias para a decisão sobre a liberação ou não do CPHD, bem como possibilitar o rastreamento para a investigação de qualquer suspeita de desvio da qualidade Os documentos devem ser elaborados, revisados e distribuídos segundo uma metodologia estabelecida pelo fabricante Os documentos devem atender a uma estrutura normativa estabelecida e formalmente proposta, com definição das responsabilidades por sua elaboração e aprovação Os documentos normativos e os registros inerentes à fabricação de CPHD são de propriedade exclusiva do fabricante, ficando à disposição da autoridade sanitária, quando solicitados Quando solicitadas formalmente pelos órgãos de vigilância sanitária competente devem os fabricantes prestar as informações ou proceder a entrega de documentos, nos prazos fixados, a fim de não obstarem a ação de vigilância e as medidas que se fizerem necessárias A documentação e registros de fabricação de CPHD devem ser arquivados, no mínimo, durante 1 ano, após o término do prazo de validade Inspeções: A inspeção é o recurso apropriado para a constatação e avaliação do cumprimento das Boas Práticas de Fabricação do Concentrado Polieletrolíticos para Hemodiálise BPFCPHD Auditorias internas devem ser realizadas periodicamente pelo fabricante, para verificar o cumprimento da BPFCPHD e suas conclusões devidamente documentadas e arquivadas Com base nas conclusões das Inspeções Sanitárias e auditorias internas devem ser estabelecidas as ações corretivas necessárias para o aprimoramento da qualidade do CPHD Os fabricantes de CPHD estão sujeitas a inspeções sanitárias para verificação do padrão de qualidade do produto com base neste Regulamento Técnico As inspeções sanitárias devem ser realizadas com base no Roteiro de Inspeção do Anexo I Os critérios para a avaliação do cumprimento dos itens do Roteiro de Inspeção, visando à qualidade e segurança do CPHD, baseiam-se no risco potencial inerente a cada item Considera-se IMPRESCINDÍVEL (I) aquele item que pode influir em grau crítico na qualidade e segurança do CPHD Considera-se NECESSÁRIO (N) aquele item que pode influir em grau menos crítico na qualidade e segurança do CPHD Considera-se RECOMENDÁVEL (R) aquele item que pode influir em grau não crítico na qualidade e segurança do CPHD Considera-se item INFORMATIVO (INF) aquele que oferece subsídios para melhor interpretação dos demais itens, sem afetar a qualidade e a segurança do CPHD O item N não cumprido após a inspeção passa a ser tratado automaticamente como I na inspeção subsequente O item R não cumprido após a inspeção passa a ser tratado automaticamente como N na inspeção subsequente, mas nunca passa a I Os itens I, N e R devem ser respondidos com SIM ou NÃO. 109
110 São passíveis de sanções aplicadas pelo órgão de Vigilância Sanitária competente, as infrações que derivam do não cumprimento dos itens qualificados como I e N no Roteiro de Inspeção, constantes do Anexo III deste Regulamento, sem prejuízo das ações legais que possam corresponder em cada caso O não cumprimento de um item I, do Roteiro de Inspeção, acarreta a suspensão imediata da atividade afetada até o seu cumprimento integral Verificado o não cumprimento de itens N, do Roteiro de Inspeção, deve ser estabelecido um prazo para adequação, de acordo com a complexidade das ações corretivas que se fizerem necessárias Verificado o não cumprimento de itens R, do Roteiro de Inspeção, o estabelecimento deve ser orientado com vistas à sua adequação. ANEXO I - ROTEIRO DE INSPEÇÃO PARA EMPRESA FABRICANTE DE CPHD 1. IDENTIFICAÇÃO DA EMPRESA a) RAZÃO SOCIAL: b) C.N.P.J.: c) NOME FANTASIA: e) Licença de funcionamento nº: afixado em local visível: ( ) sim ( ) não f) Responsável Técnico: CRF/ ( ) presente ( ) ausente h) PESSOAS CONTATADAS: 2. considerações gerais 2.1. R Os arredores do estabelecimento estão limpos e apresentam boa conservação? 2.2. R Existem fontes de poluição ou contaminação ambiental próximas ao estabelecimento? 2.3. N Existe proteção contra a entrada de roedores, insetos, aves ou outros animais? 2.4. R Existe programa formal de sanitização, desratização e desinsetização? I N F Qual a periodicidade? S N I Ã M O S N I Ã M O 110
111 N Existem registros da realização de sanitização, desratização e desinsetização? 2.5. N Os esgotos e encanamentos estão em bom estado? 2.6. N A empresa possui as plantas baixas atualizadas? 2.7. in f A empresa possui organograma? 2.8. I A empresa possui autorização de órgãos competentes, para funcionamento, referente à localização, proteção ambiental e segurança de instalações? 2.9. I N F Nº total de funcionários: (M) Nível superior: n As atribuições e responsabilidades estão formalmente descritas e entendidas pelos envolvidos? n Os funcionários são submetidos a exames médicos periódicos? in f Qual a Periodicidade? n Existem registros? r Existem sanitários em quantidade suficiente? r Estão limpos? r Existem vestiários em quantidade suficiente? r Estão limpos? in f Existe local para refeições? S N I Ã M O S N I Ã M O 111
112 in f Se não, onde os funcionários fazem suas refeições? r Os funcionários estão uniformizados? r Os uniformes estão limpos e em boas condições? n São realizados treinamentos dos funcionários? n Existem registros? R Existe gerador próprio ou outro sistema para o caso de falta de energia elétrica? in f Observações 3. ÁREA DE ARMAZENAMENTO 3.1. I N F Qual o procedimento adotado? Qual a área ocupada pelo setor em m2? 3.2. R A área tem capacidade suficiente para assegurar a estocagem ordenada e racional das matérias primas, materiais de embalagem e CPHD pronta? 3.3. N O local oferece condições de temperatura adequada para o armazenamento das matérias primas, materiais de embalagem e CPHD pronta? R Existe controle de temperatura e umidade? R Existem registros? 3.4. R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 3.5. R As paredes estão bem conservadas? S N I Ã M O S N I Ã M O 112
113 3.6. R O teto está em boas condições? 3.7. R O setor está limpo? 3.8. R A qualidade e a intensidade da iluminação são adequadas? 3.9. R A ventilação do local é suficiente e adequada? R As instalações elétricas estão em bom estado de conservação, segurança e uso? R Existem equipamentos de segurança para combater incêndios? R Os extintores estão dentro do prazo de validade? R O acesso aos extintores e mangueiras está livre? I N F Existe necessidade de câmara frigorífica e ou geladeira? R A câmara frigorífica e ou geladeira é mantida limpa sem acúmulo de gelo? N Existe controle e registro de temperatura? 3.13 R As matérias primas, materiais de embalagem e CPHD pronta estão armazenados afastados do piso e paredes, facilitando a limpeza? N Existe local para segregar matérias primas, materiais de embalagem e CPHD reprovados, recolhidos ou devolvidos? R Existem recipientes para lixo com tampa e estão devidamente identificados? N As aberturas e janelas encontram-se protegidas contra a entrada de aves, insetos, roedores e outros animais? 3.17 N As matérias primas, materiais de embalagem são inspecionados quando do seu recebimento? N As matérias primas, materiais de embalagem estão devidamente identificadas? 113
114 I As matérias primas, materiais de embalagem e CPHD e estão dentro do prazo de validade? I As matérias primas e materiais de embalagem estão acompanhados dos respectivos laudos de análises dos fornecedores, devidamente assinados pelos seus responsáveis? I O CPHD possui registro junto à agência nacional de vigilância sanitária? R Existe sistema de controle de estoque? ( ) fichas ( ) informatizado R As matérias primas e materiais de embalagem respeitam a ordem utilizandose primeiro o mais antigo? 3.20 N As matérias primas e materiais de embalagem que não são aprovados na inspeção de recebimento são rejeitados e devolvidos ou destruídos? N Existem registros? R Existem procedimentos operacionais escritos para as atividades do setor? Observações: 4. ÁGUA 4.1. I N F S N I Ã M O Qual a procedência da água utilizada na empresa? ( ) Rede Pública ( ) Poço Artesiano ( ) Semi-artesiano ( ) Outros Especificar: S N I Ã M O 114
115 4.2. I N F 4.3. I N F 4.4. I N F 4.5. I N F A empresa possui caixas d'água? Quantas? De que material? Qual o consumo médio de água da empresa? 4.6. N As instalações de água potável atendem às exigências deste Regulamento? 4.7. N É procedida limpeza da caixa d'água? I N F Qual a periodicidade? R Existem procedimentos escritos para limpeza do depósito de água potável? R Existem registros das limpezas efetuadas? 4.8. I N F I N F A água potável é submetida a algum processo de purificação? Qual? 4.9. N São realizados controles físico-químicos da água? S N I Ã M O S N I Ã M O S N I Ã M O 115
116 4.9.1 I N F I N F Qual a periodicidade? Existem registros? N São realizados controles microbiológicos da água potável? I N F I N F I N F Qual a periodicidade? Existem registros? Para que se destina a água? ( ) limpeza de material ( ) preparação de álcool a 70% ( ) preparação do CPHD Observações: 5. ÁREAS DE PREPARAÇÃO/ENVASAMENTO 5.1. I N F 5.2. in f Qual a área ocupada pelo setor em m2? As áreas destinadas à preparação/envasamento de CPHD são adequadas e suficientes ao desenvolvimento das operações, dispondo S N I Ã M O S N I Ã M O S N I Ã M O 116
117 de todos os equipamentos de forma organizada e racional? 5.3. R O piso é liso, resistente e de fácil limpeza? 5.4. R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 5.5. I N F in f Qual o n.º de funcionários que atuam na área, por turno? A circulação de pessoal nestas áreas é restrita? 5.6. N Os funcionários estão com uniforme próprio para as atividades da área de preparação/envasamento? 5.7. N Os uniformes estão limpos e em boas condições de conservação? 5.8. I Existe fórmula padrão de cada tipo de CPHD? 5.9. i Existe uma ordem de fabricação para os lotes a serem produzidos? 5.9. I Existe uma ficha técnica de fabricação que incorpore fielmente a fórmula padrão e registre as diversas etapas da fabricação do lote? N São realizados e registrados controles de processo para verificar se o CPHD está sendo fabricado conforme ficha técnica de fabricação? 5.11 N Quais os controles realizados? n Existem registros do número de lote de cada matéria prima e material de embalagem utilizados na preparação / envasamento de CPHD, indicando inclusive os seus fabricantes? S N I Ã M O S N I Ã M O 117
118 5.13. n Os recipientes utilizados para acondicionamento de CPHD atendem às especificações deste Regulamento? r É proibida a entrada de pessoas não autorizadas nos diversos setores da área de preparação / envasamento? R Existem equipamentos de Segurança para combater incêndios, atendendo à legislação específica? Observações: 6. VESTIÁRIO 6.1 I N F As áreas destinadas a vestiário são adequadas e suficientes ao desenvolvimento das operações, dispondo de todos os equipamentos de forma organizada e racional? 6.2. R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 6.3. R As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 6.4. N A iluminação é suficiente e adequada? 6.5. I N F Equipamentos Existentes: ( ) pia e torneira S N I Ã M O S N I Ã M O ( ) dispensadores para degermantes ou antissépticos ( ) toalhas descartáveis ( ) secador a ar ( ) armários para guardar uniformes ( ) cesto para despejo de roupas usadas outros. Especificar: 118
119 6.6. I N F Quais os produtos utilizados para sanitização das mãos? S NÃO I M 6.7. R Existem procedimentos escritos para a paramentação e sanitização das mãos: 6.8. Observações 10. ÁREA DE ROTULAGEM/EMBALAGEM R Existe área própria para rotulagem/embalagem? I N F Qual a área ocupada pelo setor em m2? R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? R As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? R A iluminação é suficiente e adequada? R A ventilação é suficiente e adequada? I Os rótulos apresentam todas as informações exigidas por este Regulamento? N O acondicionamento do CPHD já rotulado atende às especificações deste Regulamento? Observações 8. GARANTIA DA QUALIDADE S N I Ã M O S N I Ã M O S N 119
120 8.1. A empresa possui um sistema de Garantia da Qualidade implantado, com base nas diretrizes das BPFCPHD? 8.2. N Os procedimentos operacionais para todas as operações críticas da preparação e de controle de qualidade de CPHD estão padronizados? 8.3. N São realizadas auto-inspeções? I N F Com que frequência? N Existem registros? 8.4. N Existe um programa de treinamento para todos os funcionários? N Existem registros? 8.5. N Os pontos críticos do processo são periodicamente validados? N Existem registros? 8.6. N A documentação referente à preparação de CPHD é arquivada ordenadamente durante pelo menos 1 ano após o término do prazo de validade do produto? 8.7. N A documentação existente possibilita o rastreamento para investigação de qualquer suspeita de desvio de qualidade CPHD? 8.8. N Existem registros de reclamações referentes a desvios de qualidade de CPHD? 8.9. N Existem registros das investigações, bem como das ações corretivas? I N F Observações 9. CONTROLE DE QUALIDADE As conclusões das investigações são transmitidas por escrito ao reclamante? I Ã M O S N 120
121 9.1. in f 9.2. in f in f in f Existe laboratório de Controle de Qualidade no estabelecimento? A empresa realiza ensaios específicos com terceiros? Quais? Com quem? n Existem registros? 9.3. N O Controle de Qualidade possui pessoal técnico qualificado para exercer a função? 9.4. R O piso é liso, resistente e de fácil limpeza? 9.5. R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 9.6. R As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 9.7. R As instalações elétricas, de água e de gás estão em boas condições de uso? 9.8. n A iluminação é suficiente e adequada? 9.9. n A ventilação é suficiente e adequada? R Existem equipamentos de segurança para combater incêndios? R Os extintores estão dentro do prazo de validade? R O acesso aos extintores e mangueiras está livre? n Existem equipamentos de proteção e segurança individual (ducha, lava-olhos, óculos, etc.)? r A área de circulação está livre de obstáculos? r Existem recipientes para lixo com tampa e I Ã M O S N I Ã M O 121
122 estão devidamente identificados? n O Controle de Qualidade está equipado com aparelhos adequados para executar as análises necessárias? in f Quais são os equipamentos e aparelhos existentes? n Os equipamentos e aparelhos estão instalados de maneira adequada para corresponder às exigências do seu correto funcionamento? n Os equipamentos e aparelhos são calibrados? in f Com que frequência? n Existem registros? n Existe programa de limpeza e manutenção periódica de equipamentos e aparelhos? n Existem especificações escritas para a aquisição de matérias primas e material de embalagem? n A especificação exige o fornecimento do certificado de análise das matérias primas e materiais de embalagem? n O Controle de Qualidade monitora o cumprimento dos procedimentos de limpeza e sanitização da preparação de CPHD? n Existem métodos analíticos utilizados para a realização das análises? n São realizadas análises nos CPHD preparados, conforme estabelecido neste Regulamento? S N I Ã M O S N I Ã M O 122
123 9.25. in f n Existem registros? Em caso negativo, qual a metodologia e critério de amostragem adotado? N Existem amostras de CPHD para compor o arquivo referência durante seis meses após o término de seu prazo de validade? R Existem procedimentos operacionais escritos para o setor? Observações 10. CONCLUSÃO 11. NOME, Nº DE CREDENCIAL E ASSINATURA DOS INSPETORES 12. DATA S N I Ã M O ANEXO II - DIRETRIZES PARA REGISTRO DE CONCENTRADOS POLIELETROLÍTICOS PARA HEMODIÁLISE - CPHD 1. OBJETIVO Este Regulamento Técnico fixa os requisitos exigidos para o registro do produto caracterizado como CONCENTRADO POLIELETROLÍTICO PARA HEMODIÁLISE CPHD, considerado medicamento pela Agência Nacional de Vigilância Sanitária. 2. REFERÊNCIA 2.1. BRASIL. Decreto nº 3.181, de 23 de setembro de Regulamenta a lei 9787 que trata sobre a lei dos genéricos. Diário Oficial da União de 24 set BRASIL. Decreto n.º , de 5 de Janeiro de Regulamenta a Lei 6360, de 23 de setembro de 1976, que submete a Sistema de Vigilância Sanitária, os medicamentos, insumos farmacêuticos, drogas, correlatos, cosméticos, produtos de higiene, saneantes e outros. Diário Oficial [da República Federativa do Brasil]. Brasília, 7 jan Seção 1 P.11, Col BRASIL. Instrução Normativa n.º 01, de 30 de setembro de Trata da documentação necessária para registro. Diário Oficial da [República Federativa do Brasil]. Brasília, v.1, n.189, p , 4 out Seção 1, pt BRASIL. Portaria nº 10/SVS, de 05 de setembro de BRASIL. Portaria n.º 802, de 08 de outubro de Institui o sistema de controle e fiscalização em toda a cadeia dos produtos farmacêuticos. Diário Oficial da [República Federativa do Brasil]. Brasília, v.1, n.24-e, p. 036, 4 de fev Seção 1, pt Resolução nº 510 de 1 de outubro de 1999, Diário Oficial [da república federativa do Brasil], Brasília, 04 out Seção 1 nº 190 E p
124 3. DEFINIÇÕES 3.1. Concentrado Polieletrolítico para Hemodiálise CPHD: Concentrado de eletrólitos, com ou sem glicose, apresentado na forma sólida ou líquida para ser empregado na terapia de diálise renal, após diluição recomendada pelo fabricante e utilizando equipamento específico Dialisato: é a solução obtida após diluição do CPHD, na proporção adequada para uso. 4. ABRANGÊNCIA Exclui-se do cumprimento deste ANEXO o CPHD produzido em farmácias hospitalares e em clínicas especializadas, exclusivamente para uso próprio. 5. CONDIÇÕES GERAIS 5.1. A solicitação de registro de medicamentos caracterizados como Concentrado Polieletrolíticos para Hemodiálise - CPHD deve apresentar os seguintes documentos: Petição FP1 e PF2, em duas vias (original e cópia), indicando um similar no mercado interno, quando aplicável; Comprovante de pagamento de taxa de serviços da Vigilância Sanitária em duas vias (original e cópia), devidamente autenticadas e carimbadas, conforme estabelecido na legislação vigente; Cópia da Autorização de Funcionamento da Empresa, discriminando as funções que a empresa pode desempenhar e do Alvará Sanitário em vigor; Relatório Técnico do produto contendo: Dados Gerais: Forma farmacêutica e de apresentação. (indicar se é solução aquosa concentrada ou forma sólida pó) Características a) Quantidade: Informar a quantidade teórica proposta para apresentação; b) Aspecto: Informar se: Solução - Límpida, de incolor a levemente amarelada, odor característico, ausência de partículas visíveis a olho nu, após diluição; Sólido partículas ou granulado uniforme, sem elementos estranhos, de cor e odor característicos, facilmente solúvel em água. c) Composição: c.1) Indicar os componentes da formulação de acordo com a DCB (Denominação Comum Brasileira) (Tabela I) ou CAS (Chemical Abstract Service), e as respectiva quantidades expressas em grama por litro ou peso por peso expresso em grama; c.2) Indicar a concentração do produto após diluição expressos em meq/l e mmol/l; d) Vias de administração: via extracorpórea e) Modo de Usar: Deve ser descrito, detalhadamente, o modo particular de preparação da solução dialítica e tipo do equipamento indicado. f) Indicações do uso do concentrado; g) Contraindicações, efeitos colaterais, reações adversas do produto e da terapia dialítica; h) Restrições ou cuidados que devem ser considerados; i) Prazo de validade; j) Cuidados de conservação: indicar a temperatura em graus Celsius e outras condições de segurança necessárias para garantir a identidade, integridade, qualidade e pureza do produto, até a expiração do seu prazo de validade. 124
125 Farmacodinâmica: a) Modo de ação; b) Justificativas das doses indicadas; Produção e Controle de qualidade atendendo às diretrizes para Fabricação e Controle de Qualidade das Boas Práticas de Fabricação de CPHD: a) Fórmula completa da preparação, com todos os seus componentes especificados de acordo com a DCB ou CAS, as quantidades de cada substância expressa em grama por litro ou peso por peso expresso em grama e miliequivalentes por litro, consignando as substâncias utilizadas como veículo ou excipiente; b) Processo de Fabricação com a descrição detalhada das operações a serem realizadas incluindo esquema de obtenção da água purificada; c) Relatório descritivo detalhado do Controle de Qualidade das matérias primas e do produto acabado, descrevendo a metodologia utilizada e as especificações dos componentes da fórmula, bem como os testes de estabilidade do produto acabado e a qualidade da água Código ou convenção utilizada pela empresa para identificação dos lotes de CPHD Indicações técnicas de que não há incompatibilidade física ou química entre a embalagem adotada e os componentes de CPHD Cuidados de armazenagem e transporte; O fabricante terá que indicar as características físicas e ambientais utilizadas para armazenamento e transporte, tais como umidade, ventilação, empilhamento, dentre outros, assumindo a responsabilidade pelas especificações descritas, desde que mantidas na faixa de 10 C a 35 C Modelo de rótulo com os dizeres previsto na legislação específica e neste Regulamento Técnico, conforme o item do Anexo I Cópia do Certificado de Responsabilidade Técnica emitido pelo Conselho Regional de Farmácia Comprovante do registro de marca no Brasil ou declaração de inexistência do mesmo, firmada pelo Responsável legal e técnico da empresa, se houver; Toda a documentação deve ser apresentada no idioma português, assinada pelo representante legal da empresa, devendo a parte técnica ser firmada, conjuntamente, pelo Responsável Técnico, com o reconhecimento de firma de ambos O CPHD de procedência estrangeira deve ainda apresentar, no que couber: Registro e comprovante de comercialização no país de origem Apresentar as vantagens da fórmula proposta, com justificativa sobre ponto de vista clínico, apontando, se houver, o produto similar registrado no Brasil Declaração de órgão oficial de que o fabricante obedece as normas e requisitos de Boas Práticas de Fabricação e Controle Modelos originais de rótulos e bulas do produto importado Relatório contendo as indicações, contraindicações e advertências apresentados para o ato de Registro no País de origem, no caso de formulação que apresente componente não previsto na Tabela I Toda documentação oficial do país de origem deve ser reconhecida pelo consulado brasileiro do local de origem do produto importado. Os documentos devem ser traduzidos para o idioma português por tradutor público juramentado e assinado pelo representante legal da empresa no Brasil, devendo ser a parte técnica, também firmada, pelo representante técnico, com reconhecimento da firma de ambos Dados Complementares: Citar a descrição da substância ou componentes básicos da fórmula em Farmacopeia, formulários ou publicações. 125
126 Anexar a bibliografia sobre o produto e a literatura pertinente acompanhada, quando de origem estrangeira, de tradução integral do trabalho original, no vernáculo. A ANVISA pode solicitar trabalhos que venha a considerar necessários à avaliação da documentação científica, com duplicata para seu arquivo. TABELA I - DENOMINAÇÕES OFICIAIS DOS PRINCIPAIS ELEMENTOS USADOS NOS CONCENTRADOS POLIELETROLÍTICOS PARA HEMODIÁLISE - CPHD Denominação DCB 1.CAS Acetato de sódio (CH3COONa) Acetato de sódio triidratado (CH3COONa.3H2O) Ácido acético (CH3COOH) Ácido láctico ( C3H6O3 ) Bicarbonato de sódio (NaHCO3) Cloreto de cálcio Cloreto de cálcio diidratado (CaCl2.2H2O) Cloreto de magnésio Cloreto de magnésio hexaidratado (MgCl2.6H2O) Cloreto de potássio (KCl) Cloreto de sódio (NaCl) Glicose anidra (C6H12O6) Glicose monoidratada (C6H12O6. H2O) Lactato de sódio (C3H5NaO3) TABELA II - SOLUÇÕES CONCENTRADAS COM ACETATO OU LACTATO: MMol / L meq/l Acetato ou lactato (equivalente a bicarbonato) 32,0 45,0 32,0 45,0 Cálcio 1,0 2,0 2,0 4,0 Cloreto 90, ,0 120 Magnésio 0,25-1,2 0,5 2,4 Potássio 0,0 3,0 0,0 3,0 Sódio 130,0 145,0 130,0 145,0 Glicose 0,0 12,0 -- Soluções concentradas ácidas, SEM ADIÇÃO DE BICARBONATO: MMol / L MEq/L 126
127 Ácido Acético 2,5 10,0 2,5 10,0 Cálcio 1,0 2,0 2,0 4,0 Cloreto 90, ,0 120 Magnésio 0,25-1,2 0,5 2,4 Potássio 0,0 3,0 0,0 3,0 Sódio 80, ,0 110,0 Glicose 0,0 12,0 -- NOTA: Soluções de bicarbonato de sódio ou bicarbonato de sódio sólido, para serem adicionados imediatamente antes do uso, devem ser fornecidas em quantidades que não ultrapassem a concentração de 45 mmol/l ou 45 meq/l no produto diluído. Neste caso, deve considerar o somatório dos íons sódio na concentração final diluída das soluções concentradas ácidas. 127
128 RDC Nº 78, DE 11 DE ABRIL DE 2003 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11, inciso IV, do Regulamento da Anvisa, aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, ele o art. 111, inciso I, alínea "b", 1º do Regimento Interno aprovado pela Portaria nº 593, de 25 de agosto de 2000, republicada em 22 de dezembro de 2000, em reunião realizada em 9 de abril de 2003, considerando o disposto no Art. 7º da Lei nº 9.782, de 26 de janeiro de 1999; considerando as conclusões do Painel de Avaliação dos Hepatoprotetores, realizado em Brasília, em 3 de abril de 2002, publicadas no DOU de 27 de fevereiro de 2003, adotou a seguinte Resolução da Diretoria Colegiada e eu, Diretor Presidente- Substituto, determino a sua publicação: Art. 1º Por ocasião da primeira renovação do registro, após publicação desta Resolução, os produtos que contenham em sua formulação os seguintes princípios ativos, associados em doses fixas em uma mesma formulação ou duas ou mais apresentações em uma mesma embalagem para uso concomitante ou seqüencial, ou isolados: Adenosina; Ripason; (nome comercial), Glycyrrhiza glabra L. - (Alcaçuz); Foeniculum vulgare Hill. - (Erva-doce, Funcho); Extratos e óleos de Mentha sp. (Hortelã); Mentol; Zingiber offrcinale Rosc. - (Gengibre); Remijia ferruginea DC - (Quina mineira); Extratos de Atropa belladonna L. - (extrato de Beladona); Extratos de Cynara scolymus L. - (Alcachofra); Extratos de Rhamnus purshiana DC. - (Extrato de Cáscara Sagrada); Extratos de Solanum paniculatum L. - (extrato de Jurubeba); Vitaminas do complexo B; Pothomorphe umbellata (L.) Miq. - (elixir, extrato de Capeba); Extrato de mucosa gástrica, Extrato de pâncreas, Extrato hepático, Extrato de bile; Princípio detoxificante do fígado e que tenham indicação profilática como hepatoprotetor, devem satisfazer as seguintes exigências e condições: I - apresentar estudos clínicos de eficácia da monodroga ou da associação, publicados em revistas indexadas (Medline, Chemical Abstracts, Biosis, International Pharmaceutical Abstracts ou Biolo gical Abstracta), ou estudos clínicos de eficácia de cada princípio ativo isolado desde que se apresente justificativa técnica de que a associação é racional, ou seja, de que os efeitos de cada princípio ativo são aditivos ou sinérgicos sem apresentar maior risco ao paciente, ou II - alterar a fórmula suprimindo estes princípios ativos quando se tratar de associações ou III - alterar a indicação terapêutica com comprovada eficácia. 1º O disposto no caput deste artigo não se aplica aos pro-dutos com registro válido até 15 de abril de 2004, devendo apresentar o que dispõe esta Resolução, na segunda renovação. 2º Os produtos que não atenderem ao disposto no caput deste artigo não terão seus registros renovados já que de acordo com o Relatório do "Painel de Avaliação dos Hepatoprotetores" (vide portal da ANVISA), esses produtos expõem a população a riscos sanitários desnecessários. 3º Caso a empresa opte por alterar sua fórmula ou indicação terapêutica, poderá manter o seu registro, desde que apresente toda a documentação e comprovações segundo a legislação vigente. Art. 2º Esta Resolução entra em vigor na data da sua publicação, revogando a RDC nº 41, de 26 de janeiro de 2003, à exceção do seu anexo. 128
129 RDC Nº 05, DE 30 DE JANEIRO DE 2015 Dispõe sobre regra de transição de lágrimas artificiais e ou lubrificantes oculares da categoria de produtos para a saúde para a categoria de medicamentos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das atribuições que lhe conferem os incisos III e IV, do art. 15 da Lei n.º 9.782, de 26 de janeiro de 1999, o inciso II, e 1 e 3 do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 354 da ANVISA, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, e suas atualizações, tendo em vista o disposto nos incisos III, do art. 2º, III e IV, do art. 7º da Lei n.º 9.782, de 1999, e o Programa de Melhoria do Processo de Regulamentação da Agência, instituído por meio da Portaria nº422, de 16 de abril de 2008 em reunião realizada em 13 de maio de 2014, adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor- Presidente, determino a sua publicação: Art. 1º As lágrimas artificiais e ou lubrificantes oculares são enquadrados na categoria de medicamentos específicos. Art. 2º As petições de registro de lágrimas artificiais e ou lubrificantes oculares já protocoladas na ANVISA na categoria de produtos para a saúde até o dia serão avaliadas dentro da categoria de produtos para a saúde. Parágrafo único. As petições de registro de lágrimas artificiais e ou lubrificantes oculares protocoladas na categoria de produtos para a saúde após a data mencionada no caput deste Artigo serão indeferidas. Art. 3º Os produtos, com mesmo detentor de registro e mesma formulação, registrados em ambas as categorias terão seus registros na categoria de produtos para a saúde cancelados a partir da entrada em vigor desta Resolução. Art. 4º Os detentores de registro de lágrimas artificiais e ou lubrificantes oculares na categoria de produtos para a saúde deverão solicitar novo registro na categoria de medicamentos específicos em até 24 (vinte e quatro) meses da entrada em vigor desta Resolução. Parágrafo único. Registros de lágrimas artificiais e ou lubrificantes oculares na categoria de produtos para a saúde que vencerem em até 24 (vinte e quatro) meses, a contar da vigência desta Resolução, poderão solicitar sua revalidação como produto para saúde, mas não poderão deixar de observar o prazo final estabelecido no caput deste artigo para a solicitação de novo registro na categoria de medicamentos específicos. Art. 5º Os detentores de registro de lágrimas artificiais e ou lubrificantes oculares na categoria de produtos para a saúde que não solicitarem novo registro na categoria de medicamentos específicos no prazo de até 24 (vinte e quatro) meses da entrada em vigor desta Resolução terão seus registros cancelados. Art. 6º Esta Resolução entra em vigor na data de sua publicação. 129
130 PORTARIA N 272, DE 08 DE ABRIL DE 1998 A Secretária de Vigilância Sanitária, do Ministério da Saúde, no uso de suas atribuições legais, resolve: Art. 1º Aprovar o Regulamento Técnico para fixar os requisitos mínimos exigidos para a Terapia de Nutrição Parenteral, constante do texto Anexo desta Portaria. Art. 2º Conceder o prazo de 180 dias para que as Unidades Hospitalares e Empresas Prestadoras de Bens e/ou Serviços se adequem ao disposto nesta Portaria. Art. 3º A presente Portaria entrará em vigor na data de sua publicação, revogando-se as disposições em contrário. GONZALO VECINA NETO REGULAMENTO TÉCNICO PARA A TERAPIA DE NUTRIÇÃO PARENTERAL 1. OBJETIVO: Este Regulamento Técnico fixa os requisitos mínimos exigidos para a Terapia de Nutrição Parenteral. 2. REFERÊNCIAS: 2.1.ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (Brasil). NBR 6493 : emprego de cores para identificação de tabulação. [S.l.] : ABNT, [1994]. 2.2.ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (Brasil). NB 26 : sinalização de segurança. [S.l.] : ABNT. 2.3 BOAS práticas para fabricação de produtos farmacêuticos. Brasília: Ministério da Saúde, BOECKH, H. Vestimentas e lavanderia : apostila. [S.l.] : Sociedade Brasileira de Controle de Contaminação, BRASIL. Lei nº 8078, de 11de setembro de Código de Defesa do Consumidor. Diário Oficial [da República Federativa do Brasil], Brasília, v.128, n. 176, supl., p. 1, 12 set BRASIL. Lei nº 9431, de 6 de janeiro de Dispõe sobre a obrigatoriedade do Programa de Controle de Infecções Hospitalares nos hospitais do Brasil. Diário Oficial [da República Federativa do Brasil], Brasília, v.134, n. 4, p. 265, BRASIL. Ministério da Saúde. Portaria nº 1884, de 11 de novembro de Aprova normas para Projetos Técnicos de Estabelecimentos Assistenciais de Saúde. Diário Oficial da União [da República Federativa do Brasil], Brasília, v.132, n. 237, p , 15 dez BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 500, de 09 de outubro de Regulamento técnico de soluções parenterais de grande volume. Diário Oficial da União [da República Federativa do Brasil], Brasília, v. 135, n 197, p , 13 out BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 16, de 06 de março de Determina cumprimento das diretrizes do Guia de Boas práticas de fabricação para indústria farmacêutica e o roteiro de inspeção. Diário Oficial da União [da República Federativa do Brasil], Brasília, v.133, n. 47, p. 3176, 09 mar Martins. D. P, et at; Recomendações para o preparo de misturas estéreis. Comitê de Farmácia da Sociedade Brasileira de Nutrição Parenteral e Enteral. V. 15, nº 13, supl. 1, Jun/Agos/Set CYTRYNBAUM, H. M. Relato prático da qualificação de uma área limpa : apostila. [S.l.] : Sociedade Brasileira de Controle de Contaminação,
131 2.12 ISO : normas de gestão da qualidade e garantia da qualidade : diretrizes gerais para a aplicação das normas ISO 9001, 9002 e [S.l.] : [s.n.], ISO : sistemas de qualidade : modelo para garantia da qualidade em produção, instalação e serviços associados. [S.l.] : [s.n.], LAVAR as mãos. 1. reimp. Brasília : Ministério da Saúde, Centro de Documentação, (Série A: Normas e Manuais Técnicos). 3. DEFINIÇÕES: Para efeito deste Regulamento Técnico são adotadas as seguintes definições; 3.1. Empresas Prestadoras de Bens e/ou Serviços (EPBS): Organização capacitada, de acordo com a Legislação vigente, para oferecer bens e/ou serviços em Terapia Nutricional Equipe Multiprofissional de Terapia Nutricional: grupo formal e obrigatoriamente constituído de, pelo menos um profissional médico, farmacêutico, enfermeiro, nutricionista, habilitados e com treinamento específico para a prática da TN Farmácia: estabelecimento que atenda à legislação sanitária vigente (Federal, Estadual, Municipal), com instalações e equipamentos específicos para a preparação da Nutrição Parenteral, em área asséptica, atendendo ainda às exigências das Boas Práticas de Preparação de Nutrição Parenteral (BPPNP), conforme Anexo II Nutrição Parenteral (NP): solução ou emulsão, composta basicamente de carboidratos, aminoácidos, lipídios, vitaminas e minerais, estéril e apirogênica, acondicionada em recipiente de vidro ou plástico, destinada à administração intravenosa em pacientes desnutridos ou não, em regime hospitalar, ambulatorial ou domiciliar, visando a síntese ou manutenção dos tecidos, órgãos ou sistemas Produtos farmacêuticos: soluções parenterais de grande volume (SPGV) e soluções parenterais de pequeno volume (SPPV), empregadas como componentes para a manipulação da NP Terapia de Nutrição Parenteral (TNP): conjunto de procedimentos terapêuticos para manutenção ou recuperação do estado nutricional do paciente por meio de NP Terapia Nutricional (TN): conjunto de procedimentos terapêuticos para manutenção ou recuperação do estado nutricional do paciente por meio da Nutrição Parenteral e ou Enteral Unidade Hospitalar (UH): estabelecimento de saúde destinado a prestar assistência à população na promoção da saúde e na recuperação e reabilitação de doentes. 4. CONDIÇÕES GERAIS: 4.1. A complexidade da TNP exige o comprometimento e a capacitação de uma equipe multiprofissional para garantia da sua eficácia e segurança para os pacientes A TNP deve abranger, obrigatoriamente, as seguintes etapas: Indicação e prescrição médica Preparação: avaliação farmacêutica, manipulação, controle de qualidade, conservação e transporte Administração Controle clínico e laboratorial Avaliação final Todas as etapas descritas no item anterior devem atender a procedimentos escritos específicos e serem devidamente registradas, evidenciando as ocorrências na execução dos procedimentos As UH e as EPBS que queiram habilitar-se à prática da TNP devem contar com: Farmácia com licença de funcionamento concedida pelo órgão sanitário competente. 131
132 Equipe de Terapia Nutricional constituído por uma equipe multiprofissional de terapia nutricional (EMTN), formal e obrigatoriamente constituída de, pelo menos, um profissional de cada categoria, que cumpra efetivamente com treinamento específico para essa atividade, a saber: médico, farmacêutico, enfermeiro e nutricionista, com as respectivas atribuições descritas no Anexo I As UHS e EPBS, para exercerem as suas atividades especificas, devem cadastrar-se junto ao Órgão de Vigilância Sanitária Estadual, Municipal ou do Distrito Federal, conforme Anexo VI. NOTA: As UHS e EPBS que desenvolverem atividades de Terapia Nutricional estão sujeitas as inspeções sanitárias periódicas As UH que não possuem as condições previstas no item anterior podem contratar os serviços de terceiros, devidamente licenciadas, para a operacionalização total ou parcial, da TNP, devendo, nestes casos, formalizar um contrato por escrito. NOTA 1: Os médicos não participantes da equipe multiprofissional que queiram indicar, prescrever e acompanhar pacientes submetidos à TNP devem fazê-lo em consenso com uma equipe multiprofissional, conforme previsto no item NOTA 2: A EPBS que somente exercer a atividade de preparação da NP fica dispensada de contar com a EMTN. 4.6 As Farmácias só podem habilitar-se para a preparação da NP se preencherem os requisitos do item 4.11 e forem previamente inspecionadas. 4.7 Ao médico, de acordo com as atribuições do Anexo I, compete: indicar, prescrever e acompanhar os pacientes submetidos à TNP. 4.8 Ao farmacêutico, de acordo com as atribuições do Anexo I, compete: realizar todas as operações inerentes ao desenvolvimento, preparação (avaliação farmacêutica, manipulação, controle de qualidade, conservação e transporte) da NP, atendendo às recomendações das BPPNP, conforme Anexo II. 4.9 Ao enfermeiro, de acordo com as atribuições do Anexo I, compete: administrar NP, observando as recomendações das BPANP, conforme Anexo IV Ao nutricionista, de acordo com as atribuições do Anexo I, compete: avaliar o estado nutricional dos pacientes, suas necessidades e requerimentos As farmácias devem possuir recursos humanos, infraestrutura física, equipamentos e procedimentos operacionais que atendam às recomendações das BPPNP, conforme Anexo II É de responsabilidade da Administração da UH prever e prover os recursos humanos e materiais necessários à operacionalização da TNP Acidentes na TNP estão sujeitos às disposições previstas no Código de Defesa do Consumidor (Lei n.º 8078, de 11/09/1990) e, em especial, nos artigos 12 e 14 que tratam da responsabilidade pelo fato do produto e do serviço, independentemente da responsabilidade criminal e administrativa O descumprimento das recomendações deste Regulamento e seus anexos, sujeita os responsáveis às penalidades previstas na Legislação Sanitária vigente, sem prejuízo da cível e criminal. 5. CONDIÇÕES ESPECÍFICAS: Na aplicação deste regulamento são adotadas as seguintes condições específicas: 5.1. Indicação: O médico é o responsável pela indicação da NP A indicação da TNP deve ser precedida da avaliação nutricional periódica do paciente São candidatos à TNP os pacientes que não satisfazem suas necessidades nutricionais pela via digestiva, considerando-se também seu estado clínico e qualidade de vida Prescrição: 132
133 5.2.1.O médico é o responsável pela a prescrição da TNP A prescrição da TNP deve contemplar o tipo e a quantidade dos nutrientes requeridos pelo paciente, de acordo com seu estado mórbido, estado nutricional e requerimentos nutricionais A TNP deve atender a objetivos de curto e longo prazos. NOTA 1: Entende-se como curto prazo a interrupção ou redução da progressão das doenças, a cicatrização das feridas, a passagem para nutrição por via digestiva e a melhora do estado de desnutrição. NOTA 2: Entende-se por longo prazo a manutenção do estado nutricional normal e a reabilitação do paciente em termos de recuperação física e social Preparação: O farmacêutico é o responsável pela a preparação da NP A preparação da NP, que envolve a avaliação farmacêutica da prescrição, a manipulação, o controle de qualidade, a conservação e o transporte da NP, exige a responsabilidade e a supervisão direta do farmacêutico, devendo ser realizada, obrigatoriamente, na farmácia habilitada para este fim e de acordo com as recomendações das BPPNP, conforme Anexo II Os produtos farmacêuticos e correlatos adquiridos industrialmente para o preparo da NP, devem ser registrados no Ministério da Saúde e acompanhados do Certificado de Análise emitido pelo fabricante, garantindo a sua pureza físico-química e microbiológica, bem como o atendimento às especificações estabelecidas Os produtos farmacêuticos produzidos em farmácia de UH e/ou EPBS devem atender às Normas de Boas Práticas de Fabricação de Soluções Parenterais de Grande Volumes e/ou de Produtos Farmacêuticos A avaliação farmacêutica da prescrição da NP quanto à sua adequação, concentração e compatibilidade físicoquímica de seus componentes e dosagem de administração, deve ser realizada pelo farmacêutico antes do início da manipulação. Qualquer alteração na prescrição, que se fizer necessária, em função da avaliação farmacêutica, deve ser discutida com o médico da equipe que é o responsável por sua alteração formal Os produtos farmacêuticos e correlatos para preparação da NP devem ser previamente tratados para garantir a sua assepsia externa e inspecionados visualmente quanto à presença de partículas A manipulação da NP deve ser realizada em área classificada grau A ou B (classe 100), circundada por área grau B ou C (classe ), de acordo com as Boas Práticas para Fabricação e Controle de Produtos Farmacêuticos A manipulação da NP deve ser realizada com técnica asséptica, seguindo procedimentos escritos e validados A formulação padronizada de NP deve ter estudos de estabilidade previamente realizados para definir seu prazo de validade A formulação padronizada adicionada de qualquer outro produto, por expressa prescrição médica, transformase em uma preparação extemporânea A NP deve ser acondicionada em recipiente atóxico, apirogênico, compatível físico-quimicamente com a composição do seu conteúdo, conforme estabelecido no anexo III. O recipiente deve manter a esterilidade e apirogenicidade do seu conteúdo durante a conservação, transporte e administração e ter registro no Ministério da Saúde A NP deve ser rotulada com identificação clara do nome do paciente, composição e demais informações Legais e específicas, conforme item do Anexo II para a segurança de sua utilização e garantia da possibilidade de seu rastreamento Após a manipulação, a NP deve ser submetida à inspeção visual para garantir a ausência de partículas, precipitações, separação de fases e alterações de cor, bem como deve ser verificada a clareza e a exatidão das informações do rótulo De cada NP preparada devem ser reservadas amostras, conservadas sob refrigeração (2ºC a 8ºC), para avaliação microbiológica laboratorial e contraprova. 133
134 As amostras para avaliação microbiológica laboratorial devem ser estatisticamente representativas de uma sessão de manipulação n + 1 colhidas aleatoriamente no início e fim do processo de manipulação As amostras para contraprova de cada NP preparada, devem ser conservadas sob refrigeração (2ºC a 8ºC) durante 7 dias após o seu prazo de validade. NOTA: Somente são válidas, para fins de avaliação microbiológica, as NP nas suas embalagens originais invioladas ou suas correspondentes amostras. 5.4 Conservação: Imediatamente após o preparo e durante todo e qualquer transporte a NP deve ser mantida sob refrigeração (2ºC a 8ºC), exceto nos casos de administração imediata. 5.5 Transporte: O transporte da NP deve obedecer a critérios estabelecidos nas normas de BPPNP, conforme Anexo II O farmacêutico é responsável pela manutenção da qualidade da NP até a sua entrega ao profissional responsável pela administração e deve orientar e treinar os funcionários que realizam o seu transporte. 5.6 Administração: O Enfermeiro é responsável pela administração A administração da NP deve ser executada de forma a garantir ao paciente uma terapia segura e que permita a máxima eficácia, em relação aos custos, utilizando materiais e técnicas padronizadas A NP é inviolável até o final de sua administração, não podendo ser transferida para outro tipo de recipiente O acesso venoso para infusão da NP deve ser estabelecido sob supervisão médica ou de enfermeiro, por meio de técnica padronizada e conforme protocolo previamente estabelecido A utilização da via de acesso da NP deve ser exclusiva. A necessidade excepcional da sua utilização para administração de qualquer outra solução injetável, só pode ser feita após aprovação formal da Equipe Multiprofissional de Terapia Nutricional (EMTN). 5.7 Controle Clínico e Laboratorial: O paciente submetido à TNP deve ser controlado quanto à eficácia do tratamento, efeitos adversos e modificações clínicas que possam influir na qualidade da TN O controle do paciente em TNP deve contemplar: ingressos de nutrientes, tratamentos farmacológicos concomitantes, sinais de intolerância à NP, alterações antropométricas, bioquímicas, hematológicas e hemodinâmicas, assim como modificações em órgãos e sistemas cujas funções devem ser verificadas periodicamente Qualquer alteração encontrada nas funções dos principais órgãos e as consequentes alterações na formulação ou via de acesso da NP devem constar na história clínica do paciente. 5.8 Avaliação Final: Antes da interrupção/suspensão da TNP o paciente deve ser avaliado em relação à: a) capacidade de atender às suas necessidades nutricionais por via digestiva. b) presença de complicações que ponham o paciente em risco de vida. c) possibilidade de alcançar os objetivos propostos, conforme normas médicas e legais. 5.9 Documentação Normativa e Registros: Os documentos normativos e os requisitos inerentes à TNP são de propriedade exclusiva da UH e/ou EPBS, cabendo à fiscalização a sua avaliação (in loco), durante a inspeção sanitária Em caso de investigação por denúncias, irregularidades ou acidentes ocorridos com a TNP, a fiscalização sanitária tem o direito de solicitar cópia dos documentos e registros necessários à elucidação do problema em questão Inspeções: 134
135 As UH e as EPBS estão sujeitas a inspeções sanitárias para verificação do padrão de qualidade do Serviço de TN, com base no Anexo I, bem como o grau de atendimento às BPPNP (Anexo II) e BPANP (Anexo IV) As inspeções sanitárias devem ser realizadas com base nos Roteiros de Inspeção do Anexo V Os critérios para a avaliação do cumprimento dos itens do Roteiro de Inspeção, visando a qualidade e segurança da NP, baseiam-se no risco potencial inerente a cada item Considera-se IMPRESCINDÍVEL (I) aquele item que pode influir em grau crítico na qualidade e segurança da NP Considera-se NECESSÁRIO (N) aquele item que pode influir em grau menos crítico na qualidade e segurança da NP Considera-se RECOMENDÁVEL (R) aquele item que pode influir em grau não crítico na qualidade e segurança da NP Considera-se item INFORMATIVO (INF) aquele que oferece subsídios para melhor interpretação dos demais itens, sem afetar a qualidade e a segurança da NP O item N não cumprido após a inspeção passa a ser tratado automaticamente como I na inspeção subsequente O item R não cumprido após a inspeção passa a ser tratado automaticamente como N na inspeção subsequente, mas nunca passa a I Os itens I, N e R devem ser respondidos com SIM ou NÃO São passíveis de sanções aplicadas pelo órgão de Vigilância Sanitária competente, as infrações que derivam do não cumprimento dos itens qualificados como I e N nos Roteiros de Inspeção, constantes do Anexo V deste Regulamento, sem prejuízo das ações legais que possam corresponder em cada caso O não cumprimento de um item I, dos Roteiros de Inspeção, acarreta a suspensão imediata da atividade afetada até o seu cumprimento integral Verificado o não cumprimento de itens N, dos Roteiros de Inspeção, deve ser estabelecido um prazo para adequação, de acordo com a complexidade das ações corretivas que se fizerem necessárias Verificado o não cumprimento de itens R, dos Roteiros de Inspeção, o estabelecimento deve ser orientado com vistas à sua adequação Índice dos Anexos: Anexo I - Atribuições da Equipe Multiprofissional de Terapia Nutricional (EMTN) Anexo II - Boas Práticas de Preparação de Nutrição Parenteral (BPPNP) Anexo III - Recipientes para Nutrição Parenteral Anexo IV - Boas Práticas de Administração da Nutrição Parenteral (BPANP) Anexo V - Roteiros de Inspeção A - Identificação da Empresa e Inspeção das Atividades da EMTN B - Farmácia de Preparação de Nutrição Parenteral C - Administração de Nutrição Parenteral - Anexo VI - Ficha Cadastral das UH e EPBS para a prática da TN. ANEXO I - ATRIBUIÇÕES DA EQUIPE MULTIPROFISSIONAL DE TERAPIA NUTRICIONAL (EMTN) PARA NUTRIÇÃO PARENTERAL 1. Objetivo: Esta recomendação estabelece as atribuições da EMTN, especialmente para a prática da TNP. 2. CONSIDERAÇÕES GERAIS: 135
136 2.1 Para a execução, supervisão e a avaliação permanente em todas as etapas da TNP, é condição formal e obrigatória a constituição de uma equipe multiprofissional. 2.2 Por se tratar de procedimento realizado em pacientes sob cuidados especiais, e para garantir a vigilância constante do seu estado nutricional, a EMTN para NP, deve ser constituída de pelo menos 01(um) profissional de cada categoria, com treinamento especifico para esta atividade, a saber: médico, farmacêutico, enfermeiro e nutricionista. 2.3 A EMTN deve ter um Coordenador Técnico-Administrativo e um Coordenador Clinico, ambos integrantes da equipe e escolhidos pelos seus componentes O Coordenador Técnico-Administrativo deve, preferencialmente, possuir titulo de especialista reconhecido na área de Terapia Nutricional O Coordenador Clínico deve ser médico e preencher, pelo menos um dos critérios abaixo relacionados: Ser especialista em Terapia Nutricional, com título reconhecido Possuir Mestrado, Doutorado ou Livre Docência em área relacionada com a Terapia Nutricional. NOTA 1: O Coordenador Clinico pode ocupar, concomitantemente, a Coordenação Técnica-Administrativa desde que consensuado pela equipe. NOTA 2: As UH e EPBS situadas em regiões carentes de profissionais qualificados para exercer a função de Coordenador Clínico, de acordo com os critérios do item 2.3, têm o prazo de até 2 anos, a partir da data de publicação desta Portaria, para regularizar esta situação. 3. ATRIBUIÇÕES GERAIS DA EQUIPE MULTIPROFISSIONAL DE TN 3.1. Criar mecanismos para que se desenvolvam as etapas de triagem e vigilância nutricional, em regime hospitalar, ambulatorial ou domiciliar 3.2. Atender às solicitações de avaliação do estado nutricional do paciente, indicando, acompanhando e modificando a TN, quando necessário, e em comum acordo com o médico responsável pelo paciente, até que sejam atingidos os critérios de reabilitação nutricional preestabelecidos 3.3 Assegurar condições adequadas de indicação, prescrição, preparação, conservação, transporte e administração, controle clínico e laboratorial e avaliação final, da TNP, visando obter os benefícios máximos do procedimento e evitar riscos. 3.4 Capacitar os profissionais envolvidos, direta ou indiretamente, com a aplicação do procedimento, por meio de programas de educação continuada, devidamente registrados. 3.5 Documentar todos os resultados do controle e da avaliação da TNP visando a garantia de sua qualidade. 3.6 Estabelecer auditorias periódicas a serem realizadas por um dos membros da equipe multiprofissional, para verificar o cumprimento e o registro dos controles e avaliação da TNP. 3.7 Analisar o custo e o benefício no processo de decisão que envolve a indicação, a manutenção ou a suspensão da TNP. 3.8 Desenvolver, rever e atualizar regularmente as diretrizes e procedimentos relativos aos pacientes e aos aspectos operacionais da TNP. 4. ATRIBUIÇÕES DO COORDENADOR TÉCNICO-ADMINISTRATIVO Compete ao Coordenador Técnico-Administrativo: 4.1 Assegurar condições para o cumprimento das atribuições gerais da equipe e dos profissionais da mesma, visando prioritariamente a qualidade e efetividade da TNP. 4.2 Representar a equipe em assuntos relacionados com as atividades da EMTN. 136
137 4.3 Promover e incentivar programas de educação continuada, para todos os profissionais envolvidos na TN, devidamente registrados. 4.4 Padronizar indicadores de qualidade para a TNP, para aplicação pela EMTN. 4.5 Gerenciar aspectos técnico-administrativos das atividades da TNP. 5. ATRIBUIÇÕES DO COORDENADOR CLÍNICO: Compete ao Coordenador Clínico: 5.1. Estabelecer protocolos de avaliação, indicação, prescrição e acompanhamento da TNP Zelar pelo cumprimento das diretrizes de qualidade estabelecidas nas BPPNP e BPANP Assegurar a atualização dos conhecimentos técnico-científicos relacionados com a TNP e sua aplicação Garantir que a qualidade dos procedimentos da TNP prevaleça sobre quaisquer outros aspectos. 6. ATRIBUIÇÕES DOS PROFISSIONAIS MÉDICOS: 6.1 Indicar e prescrever a TNP. 6.2 Estabelecer o acesso intravenoso central, para a administração da NP e proceder o acesso intravenoso central, assegurando sua correta localização. 6.3 Orientar o paciente, os familiares ou o responsável legal, quanto aos riscos e benefícios do procedimento. 6.4 Participar do desenvolvimento técnico-científico relacionado ao procedimento. Garantir os registros da evolução e dos procedimentos médicos. 7. ATRIBUIÇÕES DOS PROFISSIONAIS FARMACÊUTICOS: 7.1. Selecionar, adquirir, armazenar e distribuir, criteriosamente, os produtos necessários ao preparo da NP Qualificar fornecedores e assegurar que a entrega dos produtos seja acompanhada de certificado de análise emitido pelo fabricante Avaliar a formulação da prescrição médica quanto a sua adequação, concentração e compatibilidade físicoquímica dos seus componentes e dosagem de administração Utilizar técnicas preestabelecidas de preparação da Nutrição Parenteral que assegurem: compatibilidade físicoquímica, esterilidade, apirogenicidade e ausência de partículas 7.5. Determinar o prazo de validade para cada Nutrição Parenteral padronizada, com base em critérios rígidos de controle de qualidade Assegurar que os rótulos da Nutrição Parenteral apresentem, de maneira clara e precisa, todos os dizeres exigidos no item Assegurar a correta amostragem da Nutrição Parenteral preparada para analise microbiológica e para o arquivo de referência Atender aos requisitos técnicos de manipulação da Nutrição Parenteral Participar de estudos para o desenvolvimento de novas formulações para Nutrição Parenteral Participar de estudos de farmacovigilância com base em analise de reações adversas e interações droganutrientes e nutriente-nutriente, a partir do perfil farmacoterapêutico registrado Organizar e operacionalizar as áreas e atividades da farmácia Participar, promover e registrar as atividades de treinamento operacional e de educação continuada, garantindo a atualização dos seus colaboradores, bem como para todos os profissionais envolvidos na preparação da NP Fazer o registro, que pode ser informatizado, onde conste no mínimo: a) data e hora de preparação da NP. b) nome completo do paciente e número de registro quando houver. 137
138 c) número sequencial da prescrição medica d) número de doses preparadas por prescrição e) identificação (nome e registro) do médico e do manipulador Desenvolver e atualizar regularmente as diretrizes e procedimentos relativos aos aspectos operacionais da preparação da NP Supervisionar e promover auto-inspeção nas rotinas operacionais da preparação da NP. 8. ATRIBUIÇÕES DOS PROFISSIONAIS ENFERMEIROS: Compete ao profissional enfermeiro: 8.1. Orientar o paciente, a família ou o responsável legal, quanto à utilização e controle da TN 8.2. Preparar o paciente, o material e o local para a inserção do cateter intravenoso Prescrever os cuidados de enfermagem na TN Proceder ou assegurar a punção venosa periférica, incluindo a inserção periférica central (PICC) 8.5. Assegurar a manutenção das vias de administração Receber a Nutrição Parenteral da Farmácia e assegurar a sua conservação até a sua completa administração Proceder a inspeção visual da Nutrição Parenteral antes de sua administração Avaliar e assegurar a instalação da Nutrição Parenteral observando as informações contidas no rótulo, confrontando-as com a prescrição médica Avaliar e assegurar a administração da Nutrição Parenteral, observando os princípios de assepsia Assegurar a infusão do volume prescrito, através do controle rigoroso do gotejamento, de preferência com uso de bomba de infusão Detectar, registrar e comunicar à EMTN e ou o médico responsável pelo paciente as intercorrências de qualquer ordem técnica e/ou administrativa Garantir o registro claro e preciso de informações relacionadas à administração e à evolução do paciente, quanto ao: peso, sinais vitais, balanço hídrico, glicosuria e glicemia, entre outros Efetuar e/ou supervisionar a troca do curativo do cateter venoso, com base em procedimentos preestabelecidos Participar e promover atividades de treinamento operacional e de educação continuada, garantindo a atualização de seus colaboradores Elaborar, padronizar procedimentos de enfermagem relacionados a TN Zelar pelo perfeito funcionamento das bombas de infusão Assegurar que qualquer outra droga e /ou nutriente prescritos, não sejam infundidos na mesma via de administração da Nutrição Parenteral, sem a autorização formal da EMTN. 9. ATRIBUIÇÕES DOS PROFISSIONAIS NUTRICIONISTAS: 9.1. Avaliar os indicadores nutricionais subjetivos e objetivos, com base em protocolo preestabelecido, de forma a identificar o risco ou a deficiência nutricional e a evolução de cada paciente, até a alta nutricional estabelecida pela EMTN Avaliar qualitativa e quantitativamente as necessidades de nutrientes baseadas na avaliação do estado nutricional do paciente Acompanhar a evolução nutricional dos pacientes em TN, independente da via de administração Garantir o registro, claro e preciso, de informações relacionadas à evolução nutricional do paciente Participar e promover atividades de treinamento operacional e de educação continuada, garantindo a atualização dos seus colaboradores. 138
139 ANEXO II - BOAS PRÁTICAS DE PREPARAÇÃO DE NUTRIÇÃO PARENTERAL - BPPNP 1. OBJETIVO Este regulamento fixa os procedimentos de boas práticas que devem ser observados na preparação (avaliação farmacêutica, manipulação, controle de qualidade, conservação e transporte) da NP. 2. DEFINIÇÕES Para efeito deste regulamento são adotadas as seguintes definições : 2.1. Área de dispensação: Área de atendimento ao usuário, destinada especificamente a receber, avaliar e dispensar a prescrição médica Conservação: Manutenção em condições higiênicas e sob refrigeração controlada a temperatura de 2o C à 8o C da NP, assegurando sua estabilidade físico-química e pureza microbiológica Controle de Qualidade: Conjunto de operações (programação, coordenação e execução) com o objetivo de verificar a conformidade dos produtos farmacêuticos, correlatos, materiais de embalagem e nutrição parenteral com as especificações estabelecidas Emulsão: Formulação farmacêutica que contém substâncias gordurosas em suspensão no meio aquoso, em perfeito equilíbrio, estéril e apirogênica Formulação Padronizada: Toda formulação para Nutrição Parenteral, sob prescrição médica, cujos componentes são previamente estabelecidos, com estudos de estabilidade realizados e prazo de validade definido, podendo ser empregado para diversos pacientes Manipulação: Mistura de produtos farmacêuticos para uso parenteral, realizado em condições assépticas, atendendo à prescrição médica Material de Embalagem: Recipientes, rótulos e caixas paraacondicionamento da NP Nutrição Parenteral (NP): Solução ou emulsão, composta basicamente de carboidratos, aminoácidos, lipídios, vitaminas e minerais, estéril e apirogênica, acondicionada em recipiente de vidro ou plástico, destinada à administração intravenosa em pacientes desnutridos ou não, em regime hospitalar, ambulatorial ou domiciliar, visando a síntese ou manutenção dos tecidos, órgãos ou sistemas Preparação: Conjunto de atividades que abrange a avaliação farmacêutica, manipulação, controle de qualidade, conservação e transporte da Nutrição Parenteral Preparação Extemporânea: Toda Nutrição Parenteral para início de uso em até 24 h após sua preparação, sob prescrição médica, com formulação individualizada Procedimento Asséptico: Operação realizada com a finalidade de preparar Nutrição Parenteral com a garantia da sua esterilidade Recipiente: Embalagem primária destinada ao acondicionamento da Nutrição Parenteral, de vidro ou plástico, que atendam aos requisitos estabelecidos no anexo III Sessão de Manipulação: tempo decorrido para uma ou mais manipulações da Nutrição Parenteral, sob as mesmas condições de trabalho, por um mesmo manipulador, sem qualquer interrupção do processo Solução: Formulação farmacêutica aquosa que contém carboidratos, aminoácidos, vitaminas e minerais, estéril e apirogênica Terapia de Nutrição Parenteral (TNP): Conjunto de procedimentos terapêuticos para manutenção ou recuperação do estado nutricional do paciente por meio de nutrição parenteral. 3. REFERÊNCIAS 139
140 3.1 ASHP technical assistance bulletin on quality assurance for pharmacy: prepared steril products. Am. J. Hosp. Pharm. n. 50, p , ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (Brasil). NBR 6493: emprego de cores para identificação de tabulação. [S.l.]: ABNT, [1994]. 3.3 BOAS práticas para fabricação de produtos farmacêuticos. Brasília: Ministério da Saúde, BOECKH, H. Vestimentas e lavanderia: apostila. [S.l.]: Sociedade Brasileira de Controle de Contaminação, BRASIL. Lei nº 8078, de 11de setembro de Código de Defesa do Consumidor. Diário Oficial [da República Federativa do Brasil], Brasília, v.128, n. 176, supl., p. 1, 12 set BRASIL. Decreto nº 2181, de 20 de março de Regulamenta o Código de Defesa do Consumidor. Diário Oficial [da República Federativa do Brasil], Brasília, v. 135, n. 55, p. 5644, 21 mar BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 500, de 09 de outubro de Regulamento técnico de soluções parenterais de grande volume. Diário Oficial da União [da República Federativa do Brasil], Brasília, v. 135, n 197, p , 13 out BRASIL. Ministério do Trabalho. Portaria nº 3214, de 08 de junho de NR 26: Sinalização de Segurança. Diário Oficial [da República Federativa do Brasil], Brasília, v. 116, n. 127, p.10423, 06 jul BRASIL. Ministério do Trabalho. Portaria nº 8, de 08 de maio de NR 07. Altera Norma Regulamentadora NR-7 - Programa de Controle Médico de Saúde Ocupacional. Diário Oficial [da República Federativa do Brasil], Brasília, v. 134, n. 91, p. 8202, 13 maio Martins. D. P, et at; Recomendações para o preparo de misturas estéreis. Comitê de Farmácia da Sociedade Brasileira de Nutrição Parenteral e Enteral. V. 15, nº 13, supl., Jun/Agos/Set CYTRYNBAUM, H. M. Relato prático da qualificação de uma área limpa : apostila. [S.l.] : Sociedade Brasileira de Controle de Contaminação, FARMACOPÉIA brasileira. São Paulo : Andrei, [19-] LAVAR as mãos. 1º. reimp. Brasília : Ministério da Saúde, Centro de Documentação, (Série A: Normas e Manuais Técnicos) MANUAL de processamento de artigos e superfícies em estabelecimentos de saúde. 2º. ed. Brasília : Ministério da Saúde, SÃO PAULO. Secretaria do Estado da Saúde. Centro de Vigilância Sanitária. Portaria no 4 de 18/06/97, 3.16 STERIL drug products for home use. USP/NF. v. 23, n.1206, p STERIL drug products: general information. USP/NF. v. 23, n. 1206, p , second supplement. 4. CONSIDERAÇÕES GERAIS As Boas Práticas de Preparação da Nutrição Parenteral (BPPNP) estabelecem as orientações gerais para aplicação nas operações de preparação (avaliação farmacêutica, manipulação, controle de qualidade, conservação e transporte) das NP, bem como os critérios para aquisição de produtos farmacêuticos, correlatos e materiais de embalagem. É indispensável a efetiva inspeção durante todo o processo de preparação das NP, de modo a garantir ao paciente a qualidade do produto a ser administrado Organização e Pessoal Estrutura Organizacional Toda farmácia deve ter um organograma que demonstre possuir estrutura organizacional e de pessoal suficiente para garantir que a NP por ela preparada esteja de acordo com os requisitos deste Regulamento Toda farmácia deve contar com pessoal qualificado e em quantidade suficiente para o desempenho de todas as tarefas pré-estabelecidas, para que todas as operações sejam executadas corretamente. 140
141 4.1.2 Responsabilidade As atribuições e responsabilidades individuais devem estar formalmente descritas e perfeitamente compreendidas pelos envolvidos que devem possuir autoridade suficiente para desempenhá-las O farmacêutico é o responsável pela supervisão da preparação da NP, e deve possuir conhecimentos científicos e experiência prática na atividade Compete ao farmacêutico: a) garantir a aquisição de produtos farmacêuticos, correlatos e materiais de embalagem com qualidade assegurada. b) manipular a NP de acordo com a prescrição médica e os procedimentos adequados para que seja obtida a qualidade exigida. c) aprovar os procedimentos relativos às operações de preparação e garantir a implementação dos mesmos. d) garantir que a validação do processo e a calibração dos equipamentos sejam executadas e registradas e que os relatórios sejam colocados à disposição. e) garantir que seja realizado treinamento inicial e contínuo dos funcionários e que os mesmos sejam adaptados conforme as necessidades. f) garantir que somente as pessoas autorizadas e devidamente paramentadas entrem nas áreas de manipulação Na aplicação de BPPNP é recomendável não haver sobreposição nas responsabilidades do pessoal Treinamento Deve haver um programa de treinamento, com os respectivos registros, para todo o pessoal envolvido nas atividades que podem afetar a qualidade da NP preparação, limpeza e manutenção) Os funcionários devem receber treinamento inicial e contínuo, inclusive instruções de higiene relevantes às suas atividades, além de motivação para a manutenção dos padrões de qualidade Todo pessoal deve conhecer os princípios das BPPNP Visitantes e pessoas não treinadas não devem ter acesso às áreas de manipulação. Sendo necessário, essas pessoas devem ser antecipadamente informadas sobre a conduta, higiene pessoal e uso de vestimentas protetoras e devem ser acompanhadas por pessoal autorizado Saúde, Higiene e Conduta A admissão dos funcionários deve ser precedida de exames médicos, sendo obrigatória a realização de avaliações médicas periódicas dos funcionários diretamente envolvidos na manipulação da NP, atendendo à NR nº 7 - MT - Programa de Controle Médico de Saúde Ocupacional - PCMSO Os profissionais que fazem a inspeção visual devem ser submetidas a exame oftalmológico periódico Em caso de suspeita ou confirmação de enfermidade ou lesão exposta, o profissional deve ser afastado temporária ou definitivamente de suas atividades, obedecendo a legislação específica O acesso de pessoas às áreas de preparação da NP deve ser restrito aos funcionários diretamente envolvidos Os manipuladores de NP devem atender a um alto nível de higiene e particularmente devem ser instruídos a lavar corretamente as mãos e antebraços, com escovações das unhas, utilizando antisséptico padronizado, antes de entrar na área de manipulação Todos os funcionários devem ser orientados quanto às práticas de higiene pessoal Na área de manipulação não deve ser permitido o uso de cosméticos, joias e relógios de pulso, a fim de evitar contaminação por partículas Não é permitido conversar, fumar, comer, beber, mascar ou manter plantas, alimentos, bebidas, fumo e medicamentos pessoais nas áreas de manipulação Todos os funcionários devem ser instruídos e incentivados a reportar aos seus superiores imediatos quaisquer condições relativas ao ambiente, equipamento ou pessoal que considerem prejudiciais à qualidade da NP. 141
142 Os procedimentos de higiene pessoal e a utilização de roupas protetoras devem ser exigidos a todas as pessoas para entrarem na área de manipulação, sejam elas funcionários, visitantes, administradores e inspetores Qualquer pessoa que evidencie condição inadequada de higiene ou vestuário, que possa prejudicar a qualidade da NP, deve ser afastada de sua atividade até que tal condição seja corrigida Vestuário A colocação dos uniformes e calçados, bem como a higiene preparatória para entrada nas áreas limpas devem ser realizadas em áreas especificamente designadas para vestiário e seguir procedimento recomendado para evitar contaminação Os uniformes e calçados utilizados nas áreas limpas devem cobrir completamente o corpo, constituindo barreira à liberação de partículas (respiração, tosse, espirro, suor, pele, cabelo e cosméticos) O tecido dos uniformes utilizados nas áreas limpas não deve liberar partículas ou fibras e deve proteger quanto à liberação de partículas naturais do corpo Os funcionários envolvidos na preparação da NP devem estar adequadamente uniformizados para assegurar a proteção do produto contra contaminação e os uniformes devem ser trocados a cada sessão para garantir a higiene apropriada O uniforme usado na área limpa, inclusive máscaras e luvas, deve ser esterilizado e substituído a cada sessão de trabalho Os uniformes reutilizáveis devem ser guardados separados, em ambientes fechados, até que sejam apropriadamente lavados, desinfetados e/ou esterilizados O processo de lavagem e esterilização dos uniformes deve ser validado e seguir procedimentos escritos, que não danifiquem as fibras do tecido e evitem a contaminação adicional de substâncias que possam se espalhar posteriormente Infraestrutura física Para preparação de Nutrição Parenteral, a farmácia deve atender aos requisitos quanto à estrutura deste Regulamento Técnico, e estar em conformidade com os critérios de circulações internas e externas, de instalações prediais ordinárias e especiais, de condições ambientais de conforto e de segurança Características Gerais A farmácia destinada à preparação de NP deve ser localizada, projetada e construída de forma a se adequar às operações desenvolvidas e de assegurar a qualidade das preparações, possuindo, no mínimo os seguintes ambientes: Área de manipulação Sala de limpeza e higienização dos produtos farmacêuticos e correlatos Sala de manipulação Vestiários Área de armazenamento Área de dispensação Os ambientes devem ser protegidos contra a entrada de aves, animais, insetos, roedores e poeiras Os ambientes devem possuir superfícies internas (pisos, paredes e teto) lisas, sem rachaduras, resistentes aos saneantes, que não desprendam partículas e serem facilmente laváveis As áreas e instalações devem ser adequadas e suficientes ao desenvolvimento das operações, dispondo de todos os equipamentos e materiais de forma organizada e racional, objetivando evitar os riscos de contaminação, misturas de componentes e garantir a sequência das operações Os ralos devem ser sifonados e fechados. Nota: Nas áreas de manipulação, limpeza e higienização é vedada a existência de ralos. 142
143 Os ambientes devem ser limpos e, quando apropriado, desinfetados conforme procedimentos escritos e detalhados A iluminação e ventilação devem ser suficientes para que a temperatura e a umidade relativa não deteriorem os produtos farmacêuticos e correlatos, bem como a precisão e funcionamento dos equipamentos Os vestiários, lavatórios e os sanitários devem ser de fácil acesso e suficientes para o número de funcionários. Os sanitários não devem ter comunicação direta com as áreas de manipulação e armazenamento As salas de descanso e refeitório devem ser separadas dos demais ambientes Condições Específicas As áreas de manipulação de NP deve ter dimensões que facilitem ao máximo a limpeza, a manutenção e as operações As operações de manipulação devem ser realizadas em áreas definidas especificamente para: a) limpeza e higienização dos produtos farmacêuticos, correlatos e materiais de embalagem utilizados na manipulação da NP, em área controlada grau D (classe ). b) manipulação da NP em área limpa grau A ou B (classe 100) ou sob fluxo laminar, circundada por grau C (classe ) vestiário para troca de uniformes, conforme item Nas áreas de manipulação todas as superfícies devem ser revestidas de material resistente aos agentes sanitizantes, lisas e impermeáveis para evitar acúmulo de partículas e microrganismos, possuindo cantos arredondados Nas áreas de manipulação, não devem ser usadas portas corrediças e as portas devem ser projetadas de modo a evitar superfícies que não possam ser limpas Os tetos rebaixados devem ser selados para evitar contaminação proveniente de espaço acima dos mesmos As tubulações devem ser embutidas nas paredes nas áreas de manipulação e limpeza e higienização As instalações de água potável devem ser construídas de materiais adequados e impermeáveis, para evitar infiltração e facilitar a limpeza e inspeção periódicas As instalações e reservatórios de água potável devem ser devidamente protegidas para evitar contaminações por microrganismos, insetos ou aves Condições específicas da área de Limpeza e Higienização dos produtos farmacêuticos e correlatos Esta área deve ser contígua à área de manipulação da NP, e dotada de passagem de dupla porta para a entrada de produtos farmacêuticos, correlatos e recipientes para envasamento, em condições de segurança Deve dispor de meios e equipamentos para a limpeza prévia e inspeção dos produtos farmacêuticos e correlatos, antes da sua entrada na área de manipulação Sala de Manipulação A sala destinada à manipulação de NP deve ser independente e exclusiva, dotada de filtros de ar para retenção de partículas e microrganismos, garantindo os graus recomendados (área limpa grau A ou B - classe 100 ou sob fluxo laminar em área grau C - classe ) e possuir pressão positiva A entrada na área de manipulação deve ser feita exclusivamente através de antecâmara (vestiário de barreira) Todas as superfícies da área de manipulação devem ser revestidas de material resistente aos agentes sanitizantes, serem lisas e impermeáveis, possuindo cantos arredondados Sistematicamente deve-se proceder ao controle do nível de contaminação ambiental do ar, seguindo procedimento escrito e com registros dos resultados Vestiários (antecâmaras) Os vestiários devem ser sob a forma de câmaras fechadas, preferencialmente com dois ambientes para mudança de roupa. 143
144 Devem ser ventilados, com ar filtrado com pressão inferior à da área de manipulação e superior à área externa. As portas das câmaras devem possuir um sistema de travas e de alerta visual e/ou auditivo para evitar a sua abertura simultânea Lavatórios devem possuir torneiras ou comandos do tipo que dispensem o contato das mãos para o fechamento da água. Junto ao lavatório deve existir provisão de sabão líquido ou antisséptico e recurso para secagem das mãos Área de Armazenamento A área de armazenamento deve ter capacidade suficiente para assegurar a estocagem ordenada das diversas categorias de produtos farmacêuticos, correlatos e materiais de embalagem Quando são exigidas condições especiais de armazenamento, quanto à temperatura e umidade, tais condições devem ser providenciadas e monitoradas sistematicamente, mantendo-se os seus registros Deve ser providenciada área segregada para estocagem de produtos farmacêuticos, correlatos, materiais de embalagem e NP reprovados, recolhidos ou devolvidos A conservação da NP pronta para transporte deve atender às condições estabelecidas no item Conservação e transporte - e ter assegurada as condições exigidas mediante verificações e monitoração, devidamente registradas A área de armazenamento de produtos farmacêuticos, correlatos e materiais de embalagem em quarentena deve ser devidamente demarcada e com acesso restrito às pessoas autorizadas Área de Dispensação Deve permitir a correta dispensação da NP, conforme as exigências do sistema adotado Deve ter espaço e condições suficientes para as atividades de inspeção final e acondicionamento da NP para transporte Não havendo área específica, a avaliação da prescrição médica pode ser realizada nesta área, desde que apresente uma organização compatível com as atividades realizadas. 4.3 Equipamentos e Mobiliários Localização e instalação dos equipamentos Os equipamentos devem ser localizados, projetados, instalados, adaptados e mantidos de forma a estarem adequados às operações a serem realizadas A estrutura dos equipamentos deve visar a minimização dos riscos de erro e permitir que os mesmos sejam efetivamente limpos e assim mantidos para que seja evitada a contaminação cruzada, o acúmulo de poeiras e sujeira e, de modo geral, qualquer efeito adverso sobre a qualidade da NP Os equipamentos utilizados na manipulação devem estar instalados de forma que, periodicamente, possam ser fácil e totalmente limpos A utilização de qualquer equipamento, como auxiliar do procedimento de manipulação, somente é permitido na área de manipulação se a área for validada com o equipamento em funcionamento As tubulações devem ser claramente identificadas, conforme norma específica Os instrumentos e os equipamentos do laboratório de controle devem ser adequados aos procedimentos de teste e análise adotados Os equipamentos de lavagem e limpeza devem ser escolhidos e utilizados de forma que não constituam fontes de contaminação Calibração e Verificação dos Equipamentos Os equipamentos devem ser validados e periodicamente verificados e calibrados, conforme procedimentos e especificações escritas, e devidamente registrados. 144
145 A calibração dos equipamentos só deve ser executada por pessoal capacitado, utilizando padrões rastreáveis à Rede Brasileira de Calibração, com procedimentos reconhecidos oficialmente, no mínimo uma vez ao ano Em função da frequência de uso do equipamento e dos registros das verificações dos mesmos, deve ser estabelecida a periodicidade da calibração A verificação dos equipamentos deve ser feita por pessoal treinado, empregando procedimentos escritos, com orientação específica e limites de tolerância definidos Devem haver registros das calibrações e verificações realizadas As etiquetas com datas referentes à última e à próxima calibração devem estar afixadas no equipamento Manutenção Todos os equipamentos devem ser submetidos à manutenção preventiva, de acordo com um programa formal, e corretiva, quando necessário, obedecendo a procedimentos operacionais escritos com base nas especificações dos manuais dos fabricantes Devem existir registros das manutenções preventivas e corretivas realizadas Limpeza e Desinfeção Programas e procedimentos operacionais de limpeza e desinfecção das áreas, instalações, equipamentos e materiais devem estar disponíveis ao pessoal responsável e operacional Os produtos usados na limpeza e desinfecção não devem contaminar com substâncias tóxicas, químicas, voláteis e corrosivas as instalações e os equipamentos de manipulação A água para preparação do álcool a 70% deve atender às especificações de "água para injetáveis" Os desinfetantes e detergentes devem ser monitorados quanto à contaminação microbiana Após o término do trabalho de manipulação, os equipamentos devem ser limpos e desinfetados, efetuando-se os respectivos registros desses procedimentos Antes do início do trabalho de manipulação deve ser verificada a condição de limpeza dos equipamentos e os respectivos registros É recomendável que o sistema de filtração de ar do fluxo laminar não seja desligado ao término do trabalho, a menos que, após a sua parada, seja providenciada a limpeza e desinfecção do gabinete, devendo o equipamento permanecer ligado por um período de, no mínimo, 1 hora antes do início do trabalho de manipulação Mobiliário Na área de manipulação, o mobiliário deve ser o mínimo e estritamente necessário ao trabalho aí desenvolvido O mobiliário deve ser construído de material liso, impermeável, facilmente lavável e que não libere partículas e que seja passível de desinfecção pelos agentes normalmente utilizados 4.4 MATERIAIS Para efeito desta norma, inclui-se no item materiais: produtos farmacêuticos, correlatos, materiais de embalagem e germicidas (antissépticos e desinfetantes) empregados no processo de preparação da NP Aquisição Compete ao farmacêutico o estabelecimento de critérios e a supervisão do processo de aquisição Deve haver especificação técnica detalhada de todos os materiais necessários à preparação da NP, de modo a garantir que a aquisição atenda corretamente aos padrões de qualidade estabelecidos Os materiais devem ser adquiridos somente de fornecedores qualificados quanto aos critérios de qualidade e, preferencialmente, diretamente do produtor A qualificação do fornecedor de materiais deve abranger os seguintes critérios: a) exato atendimento às especificações estabelecidas. b) os materiais devem ter registro ou serem declarados isentos de registro pelo Ministério da Saúde. 145
146 c) efetivo envio de certificado de análise dos lotes fornecidos. d) avaliação do histórico de fornecimento A qualificação de fornecedores deve ser documentada quanto ao procedimento utilizado, com os respectivos registros A quantidade adquirida dos materiais deve levar em consideração o consumo médio, o prazo de validade dos mesmos e a capacidade da área de estocagem nas condições exigidas Os recipientes adquiridos e destinados ao envasamento da NP devem ser atóxicos, apirogênicos e compatíveis físico-quimicamente com a composição do seu conteúdo, conforme anexo III Os recipientes devem manter a esterilidade, estabilidade e apirogenicidade da NP durante a sua conservação, transporte e administração Recebimento (inspeção, aprovação, reprovação) O recebimento dos materiais deve ser realizado por pessoa treinada e com conhecimentos específicos sobre os materiais e fornecedores Todos os materiais devem ser submetidos à inspeção de recebimento, devidamente documentada, para verificar a integridade da embalagem e quanto à correspondência entre o pedido, a nota de entrega e os rótulos do material recebido Qualquer divergência ou qualquer outro problema que possa afetar a qualidade do produto deve ser analisada pelo farmacêutico para orientar a devida disposição Se uma única remessa de material contiver lotes distintos, cada lote deve ser levado em consideração separadamente para inspeção e liberação Cada lote de produto farmacêutico e correlato deve ser acompanhado do respectivo certificado de análise Armazenamento Todos os materiais devem ser armazenados sob condições apropriadas, de modo a preservar a identidade e integridade dos mesmos, e de forma ordenada, para que possa ser feita a separação dos lotes e a rotação do estoque, obedecendo à regra: primeiro que entra, primeiro que sai Os materiais devem ser estocados em locais identificados, de modo a facilitar a sua localização para uso, sem riscos de troca Para os produtos farmacêuticos que exigem condições especiais de temperatura, devem existir registros que comprovem o atendimento a estas exigências Os materiais de limpeza e germicidas devem ser armazenados separadamente. 4.5 Controle do Processo de Preparação Avaliação Farmacêutica da Prescrição Cada prescrição médica deve ser avaliada quanto à viabilidade e compatibilidade dos componentes entre si e suas concentrações máximas, antes da sua manipulação Deve ser verificada a legibilidade da assinatura do médico e seu número de registro no CRM Com base nos dados da prescrição, devem ser realizados e registrados os cálculos necessários para a manipulação da formulação (peso, parâmetros dos componentes etc.) Controle Microbiológico do Processo Deve existir um programa de validação e monitoração do controle ambiental e de funcionários, para garantir a qualidade microbiológica da área de manipulação Deve ser validado e verificado, sistematicamente, o cumprimento do procedimento de lavagem das mãos e antebraços, conforme item Devem ser verificados o cumprimento dos procedimentos de limpeza e desinfecção das áreas, instalações, equipamentos e materiais empregados na manipulação da NP. 146
147 4.5.3 Manipulação (material, pessoal, processo e inspeção) Devem existir procedimentos operacionais escritos para todas as etapas do processo de manipulação Todos os produtos farmacêuticos, correlatos e recipientes devem ser limpos e desinfetados antes da entrada na área de manipulação Deve ser efetuado o registro do número sequencial de controle de cada um dos produtos farmacêuticos e correlatos utilizados na manipulação de NP, indicando inclusive os seus fabricantes O transporte dos materiais limpos e desinfetados para a sala de manipulação deve ser efetuado em bandejas ou carrinhos de aço inox através de câmara com dupla porta (pass-through) A área de manipulação da NP deve ser validada e monitorada para assegurar as recomendações estabelecidas no item Todas as superfícies de trabalho, inclusive as internas da capela de fluxo laminar, devem ser limpas e desinfetadas, com desinfetantes recomendados em Legislação do Ministério da Saúde, antes (pelo menos 30 minutos) e depois de cada sessão de manipulação Devem existir registros das operações de limpeza e desinfecção dos equipamentos empregados na manipulação Todo pessoal envolvido no processo de preparação de NP deve proceder à lavagem das mãos e antebraços e escovação das unhas, com antisséptico apropriado e recomendado em Legislação do Ministério da Saúde, antes do início de qualquer atividade na área de manipulação, após a descontaminação dos produtos farmacêuticos e correlatos ou contaminação acidental no próprio ambiente O procedimento de lavagem das mãos e antebraços deve ser validado e verificado sistematicamente Deve ser assegurado que as luvas estéreis sejam trocadas a cada 2 horas de trabalho de manipulação, e sempre que sua integridade estiver comprometida Os equipos de transferência devem ser trocados a cada sessão ininterrupta de manipulação Antes, durante e após a manipulação da NP, o farmacêutico deve conferir, cuidadosamente, a identificação do paciente e sua correspondência com a formulação prescrita O envasamento da NP deve ser feito em recipiente que atenda os requisitos deste regulamento e garanta a estabilidade físico-quimica e microbiológica da NP Rotulagem e Embalagem Devem existir procedimentos operacionais escritos para as operações de rotulagem e embalagem de NP Toda NP deve apresentar rótulo com as seguintes informações: nome do paciente, nº do leito e registro hospitalar, composição qualitativa e quantitativa de todos os componentes, osmolaridade, volume total, velocidade da infusão, via de acesso, data e hora da manipulação, prazo de validade, número sequencial de controle e condições de temperatura para conservação e transporte, nome e CRF do farmacêutico responsável A NP já rotulada deve ser acondicionada em embalagem impermeável e transparente para manter a integridade do rótulo e permitir a sua perfeita identificação durante a conservação e transporte Conservação e Transporte Toda NP deve ser conservada sob refrigeração, em geladeira exclusiva para medicamentos, com temperatura de 2ºC a 8ºC Em âmbito domiciliar, compete à EMTN verificar e orientar as condições de conservação da NP, de modo a assegurar o atendimento das exigências deste regulamento O transporte da NP deve ser feito em recipientes térmicos exclusivos, em condições pré-estabelecidas e supervisionadas pelo farmacêutico responsável pela preparação, de modo a garantir que a temperatura da NP se mantenha na faixa de 2º C a 20º C durante o tempo do transporte que não deve ultrapassar de 12 h, além de protegidas de intempéries e da incidência direta da luz solar. 147
148 4.6 Garantia da Qualidade Considerações Gerais A Garantia da Qualidade tem como objetivo assegurar que os produtos e serviços estejam dentro dos padrões de qualidade exigidos Para atingir os objetivos da Garantia da Qualidade na preparação de NP, a farmácia deve possuir um Sistema de Garantia da Qualidade (SGQ) que incorpore as BPPNP e um efetivo controle de qualidade totalmente documentado e monitorado através de auditorias da qualidade Um Sistema de Garantia da Qualidade apropriado para a preparação de NP deve assegurar que: a) as operações de preparação da NP sejam claramente especificadas por escrito e que as exigências de BPPNP sejam cumpridas. b) os controles de qualidade necessários para avaliar os produtos farmacêuticos, os correlatos, o processo de preparação (avaliação farmacêutica, manipulação, conservação e transporte) e a NP, sejam realizados de acordo com procedimentos escritos e devidamente registrados. c) os pontos críticos do processo sejam devida e periodicamente validados, com registros disponíveis. d) os equipamentos e instrumentos sejam calibrados, com documentação comprobatória. e) a NP seja corretamente preparada, segundo procedimentos apropriados. f) a NP só seja fornecida após o farmacêutico responsável ter atestado formalmente que o produto foi manipulado dentro dos padrões especificados pelas BPPNP. g) a NP seja manipulada, conservada e transportada de forma que a qualidade da mesma seja mantida até o seu uso. h) sejam realizadas auditorias da qualidade para avaliar regularmente o Sistema de Garantia da Qualidade e oferecer subsídios para a implementação de ações corretivas, de modo a assegurar um processo de melhoria contínua Controle de Qualidade da Nutrição Parenteral O Controle de Qualidade deve avaliar todos os aspectos relativos aos produtos farmacêuticos, correlatos, materiais de embalagem, NP, procedimentos de limpeza, higiene e sanitização, conservação e transporte da NP, de modo a garantir que suas especificações e critérios estabelecidos por este regulamento estejam atendidos Os produtos farmacêuticos e correlatos devem ser inspecionados no recebimento para verificar a integridade física da embalagem e as informações dos rótulos O certificado de análise de cada produto farmacêutico e correlato emitido pelo fabricante deve ser avaliado para verificar o atendimento às especificações estabelecidas Antes da desinfecção para entrada na área de manipulação, os produtos farmacêuticos e correlatos devem ser inspecionados visivelmente para verificar a sua integridade física, a ausência de partículas e as informações dos rótulos de cada unidade do lote (100%) Os procedimentos de limpeza, higiene e sanitização devem ser desenvolvidos e monitorados para verificar o cumprimento dos requisitos estabelecidos A manipulação deve ser avaliada quanto à existência, adequação e cumprimento de procedimentos padronizados e escritos A NP pronta para uso deve ser submetida aos seguintes controles: a) inspeção visual em 100% das amostras, para assegurar a integridade física da embalagem, ausência de partículas, precipitações e separação de fases. b) verificação da exatidão das informações do rótulo, atendendo ao item c) teste de esterilidade em amostra representativa das manipulações realizadas em uma sessão de trabalho, para confirmar a sua condição estéril As amostras para avaliação microbiológica laboratorial devem ser retiradas, estatisticamente, no início e fim do processo de manipulação e conservadas sob refrigeração (2ºC a 8ºC ) até a realização da análise. 148
149 As amostras para contraprova de cada NP preparada, devem ser conservadas sob refrigeração (2ºC a 8ºC) durante 7 dias após o seu prazo de validade As condições de conservação e transporte devem ser verificadas semanalmente para assegurar a manutenção das características da NP Todas as avaliações exigidas nos itens à devem ser devidamente registradas Validação O procedimento de manipulação asséptica deve ser validado para garantir a obtenção da NP estéril e com qualidade aceitável A validação deve seguir procedimento escrito que inclua a avaliação da técnica adotada, através de um procedimento simulado A validação deve abranger a metodologia empregada, o manipulador, as condições da área e dos equipamentos A validação do procedimento de manipulação deve ser realizada antes do efetivo início das atividades de uma farmácia. Sempre que houver qualquer alteração nas condições validadas conforme item , o procedimento deve ser revalidado É recomendado que, para cada manipulador, a validação técnica seja concluída com sucesso antes da sua liberação para a rotina de manipulação É recomendado que a competência técnica do manipulador seja revalidada, pelo menos, uma vez ao ano ou toda a vez que houver alteração significativa do processo m As validações e revalidações devem ser documentadas e os documentos arquivados durante 5 anos Prazo de validade Toda NP deve apresentar no rótulo um apropriado prazo de validade com indicação das condições para sua conservação A determinação do prazo de validade pode ser baseada em informações de avaliações da estabilidade físicoquímica das drogas e considerações sobre a sua esterilidade, ou através de realização de testes de estabilidade Fontes de informações sobre a estabilidade e físico-química das drogas deve incluir: referências de compêndios oficiais, recomendações dos fabricantes dos mesmos e pesquisas publicadas Na interpretação das informações sobre a estabilidade das drogas devem ser considerados todos os aspectos de acondicionamento e conservação Os estudos de estabilidade devem ser realizados de acordo com, uma programação escrita que abranja: a) Descrição completa da NP b) Indicação de todos os parâmetros e métodos de teste que evidenciem a estabilidade da NP quanto às suas características físicas, pureza, potência, esterilidade e apirogenicidade. c) Indicação do tempo e das condições especiais de conservação, transporte e administração, abrangidos pelo estudo. d) Registro de todos os dados obtidos, com avaliação e conclusão dos estudos Ocorrendo mudança significativa no procedimento de preparação, preparador, equipamentos, produtos farmacêuticos, correlatos e materiais de embalagem, que possa afetar a estabilidade e, portanto alterar o prazo de validade da NP, deve ser realizado novo estudo de estabilidade Reclamações Toda reclamação referente ao desvio de qualidade da NP ou das atividades relacionadas a TNP deve ser feita por escrito e analisada pela EMTN A reclamação de qualidade da NP deve incluir nome e dados pessoais do paciente, da unidade hospitalar ou do médico, nome do produto, número sequencial de controle da NP, natureza da reclamação e responsável pela reclamação. 149
150 A EMTN, ao analisar a reclamação deve estabelecer as investigações a serem efetuadas e os responsáveis pelas mesmas As investigações e suas conclusões, bem como as ações corretivas implantadas, devem ser registradas A EMTN, com base nas conclusões da investigação, deve prestar esclarecimentos por escrito ao reclamante Em caso de não ser necessária a investigação, o registro deve incluir a razão pela qual a investigação foi considerada desnecessária Documentação A documentação tem como objetivo definir as especificações de todos os materiais, de embalagem, produtos farmacêuticos e correlatos, os métodos de manipulação e controle da NP, a fim de garantir que todo o pessoal envolvido saiba decidir o que, como e quando fazer A documentação deve garantir a disponibilidade de todas as informações necessárias para a decisão sobre a liberação ou não de uma NP preparada, bem como possibilitar o rastreamento para a investigação de qualquer suspeita de desvio da qualidade Os documentos devem ser elaborados, revisados e distribuídos segundo uma metodologia estabelecida Os documentos devem atender a uma estrutura normativa estabelecida e formalmente proposta, com definição das responsabilidades por sua elaboração e aprovação A documentação/registros da NP devem ser arquivadas durante 5 anos Inspeções A farmácia das UH e da EPBS de TNP, estão automaticamente sujeitas à Inspeção Sanitária de acordo com Anexo V - Roteiros de Inspeção, cujas conclusões devem ser devidamente documentadas A inspeção é o recurso apropriado para a constatação e avaliação do cumprimento das Boas Práticas de Preparação de Nutrição Parenteral (BPPNP) Auditorias internas devem ser realizadas periodicamente na Farmácia, para verificar o cumprimento das BPPNP e suas conclusões devidamente documentadas e arquivadas Com base nas conclusões das Inspeções Sanitárias e auditorias internas devem ser estabelecidas as ações corretivas necessárias para o aprimoramento da qualidade da Terapia de Nutrição Parenteral. ANEXO III - RECIPIENTES PARA NUTRIÇÃO PARENTERAL 1. Objetivo: Este regulamento técnico fixa os requisitos mínimos relativos aos aspectos físicos, químicos e biológicos dos recipientes para envase da Nutrição Parenteral (NP). 2. Definições: 2.1 Alça de sustentação: Alça localizada na extremidade oposta aos tubos de transferência ou de conexão. 2.2 Embalagem primária: recipiente de plástico ou vidro com tampa de elastômero, empregado para o envasamento da NP. 2.3 Embalagem secundária: Materiais empregados para o acondicionamento da embalagem primária. 2.4 EVA: Poli (etileno-acetato de vinila). 2.5 Número de Lote: Designação impressa em cada unidade do recipiente constituída de combinações de letras, números ou símbolos. 2.6 Prazo de Validade: Tempo (em meses ou anos) durante o qual o recipiente mantém-se dentro dos limites especificados e com as mesmas propriedades e características que possuía quando da época da sua fabricação. 3. Referências: 150
151 3.1 BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 500, de 09 de outubro de Regulamento técnico de soluções parenterais de grande volume. Diário Oficial da União [da República Federativa do Brasil], Brasília, v. 135, n 197, p , 13 out BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 3, de 07 de fevereiro de Estabelece que todo produto correlato estéril deve ser registrado e conter, em rótulo, o número, o número do lote, a data da esterilização, o processo de esterilização usado e o prazo máximo de validade da esterilização. Diário Oficial da União [da República Federativa do Brasil], Brasília, v. 124, n. 28, p. 2326, 12 fev BRASIL. Ministério da Saúde. Secretaria de Vigilância Sanitária. Portaria nº 4, de 07 de fevereiro de Define o material médico-hospitalar de uso único, descartáveis, e proíbe seu reaproveitamento em todo o Território Nacional, em qualquer tipo de serviço de saúde. Diário Oficial da União [da República Federativa do Brasil], Brasília, v. 124, n. 28, p. 2326, 12 fev FARMACOPÉIA americana. ed. vigente. [S.l.] : [s.n.], FARMACOPÉIA brasileira. ed. vigente. [S.l.] : [s.n.], FARMACOPÉIA europeia. 3. ed. [S.l.] : [s.n.], Considerações Gerais: 4.1. Os recipientes para envase de Nutrição Parenteral (NP) podem ser de vidro e/ou plástico, desde que atendam aos requisitos estabelecidos neste Regulamento Os recipientes de vidro para envase da NP devem atender aos requisitos estabelecidos na Portaria SVS-MS n.º 500/ Os recipientes plásticos para envase da NP devem ser transparentes, sem pigmentos ou corantes, flexíveis, atóxicos, estéreis, apirogênicos, resistentes a vazamento, queda e pressão e compatíveis com a NP sob condições normais de estocagem O plástico utilizado para fabricação do recipiente deve ser o poli(etileno-acetato de vinila) - EVA, ou outros que venham a ser aprovados pelo Ministério da Saúde Os recipientes plásticos devem ser estáveis, biológica, química e fisicamente em relação ao seu conteúdo durante a validade da NP e não devem permitir a entrada de microrganismos Os recipientes plásticos não devem liberar substâncias acima dos limites especificados, para os testes propostos Os recipientes plásticos devem apresentar impresso o número de lote, que permita o rastreamento das matériasprimas, processo e componentes utilizados na sua fabricação Cada lote de recipiente fornecido deve ser acompanhado do respectivo Certificado de Análise emitido pelo fabricante, comprovando o atendimento às especificações deste Regulamento O recipiente plástico deve ser compatível com o agente esterilizante empregado e não apresentar resíduos do processo de esterilização, conforme norma específica O projeto do recipiente plástico deve ser tal que assegure a manipulação, conservação, transporte e administração da NP, sem influir na sua preservação e sem risco de contaminação por microrganismos Os tubos e conectores dos recipientes plásticos destinados à preparação, utilização e administração da NP devem garantir a adaptação e vedação com os correlatos utilizados na terapia da NP Tubo com conexão para adição de produtos farmacêuticos apresentando um conector para equipo de transferência, com protetor e pinça corta-fluxo irreversível Tubo apresentando um conector, que permita a conexão do equipo de infusão com segurança, também de vazamento, durante a administração da NP, à prova de violação e fabricado de tal modo que, qualquer tentativa de manipulação seja facilmente identificada. 151
152 Tubo com conexão para adição suplementar de medicamentos apresentando conector protegido à prova de violação e deve ser dotado de elastômero auto-cicatrizante, usado apenas de acordo com o item do Regulamento Técnico O recipiente plástico deve ter alça de sustentação que não interfira na sua utilização e resistência suficiente para suportar o seu peso nominal durante todo o período de utilização O local de etiquetagem deve deixar parte do recipiente visível e livre de marcações, para permitir a inspeção final do produto O texto de identificação do recipiente deve estar conforme a legislação vigente, na língua portuguesa, contendo obrigatoriamente, as seguintes informações: a.identificação do fabricante; b.nome do responsável técnico; c.volume nominal; d.escala graduada; e.número do lote; f.data de fabricação; g.prazo de validade O recipiente plástico deve apresentar invólucro protetor que permita a sua esterilização e garanta a manutenção da esterilidade do recipiente durante todo o prazo de validade e nas condições recomendadas pelo fabricante O recipiente plástico deve ser registrado no Ministério da Saúde. 5. Condições Específicas: 5.1. Requisitos Físicos: Controle visual: os recipientes plásticos devem ser observados quanto ao seu aspecto geral, não devendo apresentar: a) falhas na fabricação (fissuras, rebarbas internas, solda fraca). b) inclusões de materiais internos e externos. c) partículas estranhas. d) sistema de fechamento deficiente. e) falta de uniformidade na soldagem com conectores e tubos Distribuição do material: O recipiente plástico deve apresentar espessura uniforme e que comprovadamente sirva como barreira física à penetração de microrganismos e perdas excessivas de vapor d'água Transparência: O recipiente plástico deve ter uma transparência que possibilite a verificação, contra a luz, dos aspectos de limpidez da NP nele envasados, permitindo a observação de partículas, turbidez e mudança da cor Firmeza e estanqueidade das conexões: As conexões do recipiente plástico cheio com os equipos deve garantir uma perfeita conexão de modo que não haja vazamento e que os equipos permaneçam perfeitamente conectados, quando sustentados e submetidos à tração Resistência da alça de sustentação: A alça de sustentação deve permitir a utilização do recipiente cheio, pendurado, nas condições de manipulação e administração, sem apresentar sinais de ruptura ou deformação Resistência ao impacto: O recipiente plástico contendo a NP deve resistir ao impacto sem apresentar ruptura, fissura ou vazamento. 152
153 Estanqueidade do local de adição suplementar de medicamentos: O local de adição deve permanecer estanque depois da punção e retirada da agulha com 0,6 mm de diâmetro externo Soldagem dos tubos de conexões com o recipiente: Os pontos de junção dos tubos com o recipiente contendo a NP não devem apresentar vazamento quando submetido à pressão de 20kPa, durante 15 segundos Requisitos Químicos: O recipiente plástico para envase da NP deve atender aos requisitos da Farmacopeia Europeia: 3a Edição Nota: O recipiente plástico de cloreto de polivinila (PVC) não pode ser utilizado para envase da NP, contendo ou não lipídeos em sua composição Requisitos Biológicos: Impermeabilidade à microrganismos: Durante a manipulação, conservação, transporte e administração, o recipiente plástico deve garantir a esterilidade da NP nele contido Toxicidade: O recipiente plástico não deve liberar para a NP nele contida, substâncias capazes de exercerem efeitos tóxicos. Os componentes de tintas de impressão não devem atravessar as paredes do recipiente Substâncias Pirogênicas: O recipiente plástico não deve liberar, para a NP nele contida, substâncias capazes de exercerem efeitos pirogênicos Esterilidade: o recipiente plástico deve ser estéril. 6. Métodos de Ensaio para os Recipientes Plásticos 6.1. Ensaios Físicos: Para os ensaios físicos dos recipientes plásticos para envase da NP deve ser adotada a metodologia descrita no anexo E da Portaria SVS/MS nº 500/97 para os itens correspondentes aos requisitos físicos indicados neste Regulamento Ensaios Químicos: Para os ensaios químicos dos recipientes plásticos para envase de NP deve ser adotada a metodologia descrita na Farmacopeia Europeia: 3ª Ed Ensaios Biológicos: Para os ensaios biológicos deve ser adotada a metodologia descrita no anexo M da Portaria SVS-MS nº 500/97, para os itens correspondentes aos requisitos biológicos indicados neste Regulamento. 7. Aceitação e Rejeição: Os recipientes plásticos devem ser aceitos desde que atendam às exigências deste Regulamento. Caso contrário, devem ser rejeitados. ANEXO IV - BOAS PRÁTICAS DE ADMINISTRAÇÃO DA NUTRIÇÃO PARENTERAL - BPANP 1. Objetivo Este Regulamento fixa os procedimentos de Boas Práticas na Administração da Nutrição Parenteral (BPANP), que devem ser observados pela equipe de enfermagem, assegurando que a operacionalização da mesma seja realizada de forma correta. 2. Definições Para efeito deste Regulamento são adotadas as seguintes definições: 2.1 Conservação: é a manutenção em condições higiênicas e sob refrigeração controlada à temperatura de 2ºC a 8ºC da NP, assegurando sua estabilidade físico química e pureza microbiológica. 153
154 2.2 Emulsão: formulação farmacêutica que contém substâncias gordurosas em suspensão no meio aquoso, em perfeito equilíbrio, estéril e apirogênica. 2.3 Local de manuseio da NP: bancada, balcão ou mesa utilizada para o manuseio da Nutrição Parenteral antes de sua administração, localizada em área compatível com as condições de higiene e assepsia necessárias à manutenção da qualidade da NP. 2.4 Manuseio: operação de assepsia do recipiente da Nutrição Parenteral e adaptação do equipo indicado em condições de rigorosa assepsia, para proceder à sua administração. 2.5 Nutrição Parenteral (NP): solução ou emulsão, composta basicamente de carboidratos, aminoácidos, lipídios, vitaminas e minerais, estéril e apirogênica, acondicionada em recipiente de vidro ou plástico, destinado à administração intravenosa em pacientes desnutridos ou não, em regime hospitalar, ambulatorial ou domiciliar, visando a síntese ou manutenção dos tecidos, órgãos ou sistemas. 2.6 Recipiente: embalagem primária destinada ao acondicionamento de solução ou emulsão para Nutrição Parenteral, de vidro ou plástico. 2.7 Solução: formulação farmacêutica aquosa que contém carboidratos, aminoácidos, vitaminas e minerais, estéril e apirogênico. 2.8 Terapia de Nutrição Parenteral (TNP): conjunto de procedimentos terapêuticos para manutenção ou recuperação do estado nutricional do paciente por meio de nutrição parenteral. 3. Referências 3.2 Ministério da Saúde. Portaria Ministerial no 930/92 Normas para Controle de Infecções Hospitalares. Brasília. Centro de Documentação Ministério da Saúde - Centro de Documentação, Série A: Normas e Manuais Técnicos: Lavar as Mãos - Informações para Profissionais de Saúde. 1ª impressão Ministério da Saúde - Manual de Processamento de Artigos e Superfícies em Estabelecimentos de Saúde. Brasília - 2ª Edição Stier, C.J.N - Rotinas em Controle de Infecção Hospitalar - Ed. Netsul - Curitiba Lei n.º 7498, de 25 de Junho de 1.986, regulamentada pelo Decreto-Lei n.º , de 08 de Junho de Resolução do Conselho Federal de Enfermagem n.º 146, de 01 de Junho de Resolução do Conselho Federal de Enfermagem n 162, de 14 de maio de Resolução do Conselho Federal de Enfermagem n 168, de 6 de outubro de Resolução do Conselho Federal de Enfermagem n.º 186, de 20 de Julho de Resolução do Conselho Federal de Enfermagem n.º 189, de 25 de Março de Considerações Gerais As BPANP estabelecem os critérios a serem seguidos pelas Unidades Hospitalares (UH) ou Empresas Prestadoras de Bens e Serviços (EPBS) na administração da NP, em nível hospitalar, ambulatorial ou domiciliar. 4.1 Organização de Pessoal A UH/EPBS deve contar com um quadro de pessoal de enfermagem, qualificado e em quantidade que permita atender aos requisitos deste regulamento Responsabilidade A Equipe de Enfermagem envolvida na administração da NP é formada pelo Enfermeiro, Técnico de Enfermagem e Auxiliar de Enfermagem, tendo cada profissional suas atribuições dispostas em Legislação específica O Enfermeiro é o coordenador da equipe de enfermagem cabendo-lhe as ações de planejamento, organização, coordenação, execução e avaliação de serviços de enfermagem e treinamento de pessoal. 154
155 O Enfermeiro deve participar do processo de seleção, padronização, licitação e aquisição de equipamentos e materiais utilizados na administração e controle da TNP O Enfermeiro é o responsável pela administração da NP e prescrição dos cuidados de enfermagem em nível hospitalar, ambulatorial e domiciliar Ao atendente de enfermagem e equivalentes é vedada a assistência direta ao paciente em TNP. Suas atribuições estão previstas em Legislação específica Treinamento O Enfermeiro da EMTN deve participar e promover atividades de treinamento operacional e de educação continuada, garantindo a capacitação e atualização de seus colaboradores A equipe de enfermagem envolvida na administração da NP deve conhecer os princípios das Boas Práticas de Administração da NP O treinamento da equipe de enfermagem deve seguir uma programação preestabelecida e adaptada às necessidades do serviço, com os devidos registros em livro próprio O Enfermeiro da EMTN deve regularmente desenvolver, rever e atualizar os procedimentos relativos ao cuidado com o paciente em TNP Saúde, Higiene e Conduta A admissão dos funcionários deve ser precedida de exames médicos, sendo obrigatório a realização de avaliações periódicas, conforme estabelecido na NR n.º 7 do Ministério do Trabalho Havendo suspeita ou confirmação de enfermidade ou lesão exposta, o profissional deve ser afastado temporária ou definitivamente de suas atividades, obedecendo à legislação específica A equipe de enfermagem deve atender a um alto nível de higiene sendo orientados para a correta lavagem das mãos e retirada de jóias e relógios antes de operacionalizar a administração da NP Todos os funcionários devem ser instruídos e incentivados a reportar aos seus superiores imediatos quaisquer condições relativas ao ambiente, equipamento ou pessoal que considerem prejudiciais à qualidade da NP Qualquer pessoa que evidencie condição inadequada de higiene ou vestuário que possa prejudicar a qualidade da NP, deve ser afastada de sua atividade até que tal condição seja corrigida A conduta da equipe de enfermagem deve ser pautada pelos preceitos éticos em relação a sua atividade profissional, bem como ao paciente e sua família. 5. EQUIPAMENTOS E MATERIAIS 5.1 A utilização de bombas infusoras deve ser efetuada por profissional devidamente treinado. 5.2 A UH/EPBS deve garantir a disponibilidade de bombas infusoras adequadas às faixas etárias, em número suficiente, calibradas e com manutenção periódica realizadas por empresa qualificada. 5.3 As bombas infusoras devem ser periodicamente limpas e desinfetadas conforme instrução escrita estabelecida pela Comissão/Serviço de Controle de Infecção Hospitalar ( CCIH /SCIH). 5.4 Antes do início da sua utilização, as bombas infusoras devem ser cuidadosamente verificadas quanto às suas condições de limpeza e funcionamento. 5.5 Devem existir registros das operações de limpeza, desinfecção, calibração e manutenção das bombas infusoras. 5.6 A UH/ EBPS é responsável pela disponibilidade e utilização de equipos de infusão específicos para cada caso, com qualidade assegurada e em quantidade necessária à operacionalização da administração da NP. 6. Operacionalização da Administração Todos os procedimentos pertinentes à administração da NP devem ser realizadas de acordo com procedimentos operacionais escritos e que atendam às diretrizes deste Regulamento. 155
156 6.1 Preparo do Paciente e Acesso Intravenoso Orientar o paciente e sua família quanto à: a) Terapia, seus objetivos e riscos, ressaltando a importância da participação dos mesmos durante todo o processo. b) Via de administração da NP, técnica de inserção do cateter e as possíveis intercorrências que possam advir, enfatizando que a comunicação destas imediatamente à equipe de enfermagem, possibilita que as providências sejam tomadas em tempo hábil A equipe de enfermagem deve facilitar o intercâmbio entre os pacientes submetidos à NP e suas famílias, visando minimizar receios e apreensões quanto à terapia implementada O Enfermeiro deve participar da escolha da via de administração da NP, em consonância com o médico responsável pelo atendimento ao paciente É responsabilidade do Enfermeiro estabelecer o acesso intravenoso periférico, incluindo a inserção periférica de localização central (PICC) para administração da NP. NOTA: O acesso intravenoso de localização central por inserção periférica (PICC) deve ser realizado de preferência no Centro Cirúrgico, utilizando-se técnica asséptica e material estéril, obedecendo-se a procedimento estabelecido em consonância com a CCIH/SCIH O Enfermeiro deve assessorar o médico na instalação do acesso intravenoso central, que deve ser realizado de preferência no Centro Cirúrgico, utilizando-se técnica asséptica e material estéril, obedecendo-se a procedimento estabelecido em consonância com a CCIH/SCIH Na inserção do catéter venoso central, compete ao Enfermeiro: a) Providenciar o material necessário ao procedimento; b) Manter o material de reanimação cardiorrespiratória, para casos de emergência, em condições de uso; c) Preparar a região onde será inserido o catéter. d) Posicionar o paciente para o procedimento, colaborando na antissepsia da região. e) Observar sinais de desconforto que possam evidenciar complicações de ordem mecânica, intervindo de maneira correta e em tempo hábil; f) Manter a permeabilidade do catéter; g) Fazer o curativo no local de inserção do catéter, de forma a garantir sua manutenção e fixação, anotando a data do procedimento, data da troca do curativo e nome do profissional que o realizou; h) Encaminhar o paciente para exame radiológico visando a confirmação da localização do catéter. 6.2 Local de Manuseio da NP O local utilizado no manuseio da NP, deve ser revestido de material liso e impermeável, para evitar o acúmulo de partículas e microrganismos e ser resistente aos agentes sanitizantes O local de manuseio da NP deve estar organizado e livre de qualquer outra medicação e material estranhos à NP A área ao redor do local de manuseio da NP deve ser mantida em rigorosas condições de higiene Proceder à limpeza e desinfecção da área e do local de manuseio da NP conforme procedimento estabelecido pela CCIH/SCIH. 6.3 Recebimento da NP É da responsabilidade do Enfermeiro, o recebimento da NP entregue pela farmácia No recebimento o Enfermeiro deve: a) Observar a integridade da embalagem, presença de partículas, precipitações, alteração de cor e separação de fases da NP. 156
157 b) Realizar a inspeção de recebimento, verificando no rótulo: o nome do paciente, no. do leito e registro hospitalar, data e hora da manipulação, composição, osmolaridade e volume total, velocidade de infusão e prazo de validade, nome do farmacêutico responsável e registro no órgão de classe Verificada alguma anormalidade, a NP não deve ser administrada. O farmacêutico responsável pela preparação deve ser contactado e os recipientes devolvidos à farmácia. O Enfermeiro deve registrar o ocorrido em livro próprio e assinar de forma legível, anotando seu número de registro no órgão de classe. 6.4 Conservação da NP Toda NP deve ser mantida sob refrigeração em geladeira exclusiva para medicamentos, mantendo-se a temperatura entre 2ºC e 8ºC A geladeira utilizada para conservação da NP deve ser limpa, conforme procedimentos estabelecida pela CCIH/SCIH Administração da NP Retirar a NP da geladeira com antecedência necessária para que a mesma atinja a temperatura ambiente, recomendada para a sua administração Observar a integridade da embalagem, presença de partículas, precipitações, alteração de cor e separação de fases da NP Conferir no rótulo o nome do paciente, número do leito e registro hospitalar, data e hora da manipulação, composição, osmolaridade e volume total, velocidade de infusão e prazo de validade Proceder à correta lavagem das mãos, retirando jóias e relógio, antes de prosseguir na operacionalização da administração da NP Confirmar a localização do catéter venoso e sua permeabilidade, antes de iniciar a infusão da NP Adaptar o equipo de infusão adequado ao recipiente contendo a NP Evitar a exposição da NP à incidência direta de luz. Nota: Cuidado especial deve ser observado quando a NP for administrada à recém-nascido submetido à fototerapia Manter o recipiente da NP e o equipo de infusão afastados de fontes geradoras de calor A via de acesso utilizada para a administração da NP é exclusiva. É vedada a sua utilização para outros procedimentos. Casos excepcionais devem ser submetidos à avaliação da EMTN Recomenda-se a utilização de filtros na linha de infusão da NP, para maior segurança do procedimento É vedada a transferência da NP para outro recipiente Proceder à antissepsia das conexões do catéter na troca do equipo, solução indicada pela CCHI/SCIH Administrar a NP de modo contínuo, cumprindo rigorosamente o prazo estabelecido para infusão. É vedado à equipe de enfermagem, a compensação do volume no caso de atraso ou infusão rápida. Recomenda-se a utilização de bombas infusoras adequadas à faixa etária Garantir que a via de acesso da NP seja mantida, conforme prescrição médica ou rotina preestabelecida pelo serviço, no caso de ocorrer descontinuidade na administração Substituir o curativo da região de inserção do cateter venoso de localização central de forma a manter sua fixação e manutenção. A troca do curativo deve ser feita de acordo com as diretrizes estabelecidas pela CCIH/SCIH. 6.6 Monitorização do Paciente Proporcionar ao paciente uma assistência de enfermagem humanizada, procurando ouvir suas queixas e expectativas em relação à Terapia Nutricional, tranquilizando-o e mantendo-o informado de sua evolução Adotar medidas de higiene e conforto que proporcione bem estar ao paciente, estimulando o banho diário, higiene oral, mudança de decúbito com frequência ou deambulação Observar sinais e sintomas de complicações mecânicas, metabólicas e infecciosas, comunicando ao médico responsável pelo atendimento ao paciente e registrando na evolução de enfermagem e livro de relatório da unidade. 157
158 6.6.4 Pesar diariamente o paciente, sempre que possível, preferencialmente no mesmo horário e na mesma balança e com o mesmo tipo de vestimenta Verificar os sinais vitais, conforme prescrição ou rotina preestabelecida pelo serviço Realizar a glicemia capilar e a glicosúria de resultado imediato, conforme prescrição ou rotina preestabelecida pelo serviço Realizar o balanço hídrico rigoroso O Enfermeiro deve assegurar a realização dos exames clínicos e laboratoriais solicitados, atendendo rigorosamente tempo e prazo. 6.7 Registros Registrar em livros e impressos próprios, todos os dados e ocorrências referentes ao paciente e sua evolução, assim como intercorrências com a NP É da responsabilidade do Enfermeiro assegurar que todas as ocorrências e dados referentes ao paciente e à TNP sejam registrados de forma correta, garantindo a disponibilidade de informações necessárias à avaliação do paciente e eficácia do tratamento. 6.8 Inspeções As unidades que administram NP estão automaticamente sujeitas à Inspeção Sanitária de acordo com o Anexo V - Roteiros de Inspeção, cujas conclusões devem ser documentadas A Inspeção é o procedimento apropriado para avaliação do cumprimento das BPANP Auditorias Internas devem ser realizadas periodicamente para avaliar as atividades de administração da NP, verificar o cumprimento das BPANP e suas conclusões documentadas e arquivadas Com base nas conclusões das Inspeções Sanitárias e Auditorias Internas, devem ser estabelecidas as ações corretivas necessárias para o aprimoramento da qualidade da TNP. ANEXO V A - ROTEIRO PARA IDENTIFICAÇÃO DA EMPRESA E INSPEÇÃO das atividades da EMTN A1 - IDENTIFICAÇÃO DA EMPRESA a) RAZÃO SOCIAL: b) C.G.C.: / / c) NOME FANTASIA: d) ENDEREÇO: CEP: - BAIRRO: MUNICÍPIO: UF: 821 FONE: ( ) FAX: ( ) E.MAIL: e) TIPO DE EMPRESA UNIDADE HOSPITALAR (UH) EMPRESA PRESTADORA DE BENS E SERVIÇOS 158
159 (EPBS) A2 INSPEÇÃO DAS ATIVIDADES DA EMTN SIM NÃO 1. INF A UH/EPBS utiliza Terapia de Nutrição Parenteral? (Caso negativo, é desnecessário preencher este questionário) 2. INFA UH/EPBS conta com Farmácia para preparação de NP? ( Caso negativo, passar para o ítem 9 ) 3. INFA UH/EPBS conta com uma EMTN, formalmente constituída? (Caso negativo, passar para o item 9) 4. I Existe ato formal de constituição da EMTN? 5. INF Qual a composição da EMTN? COORDENADOR CLÍNICO COORDENADOR TÉCNICO ADMINISTRATIVO MÉDICO FARMACÉUTICO ENFERMEIRO NUTRICIONISTA OUTROS, ESPECIFICAR 6. INF Os membros da EMTN possuem título de especialista ou treinamento específico? MEMBROS NÃO/TÍTULO ESP./TREINAMENTO ESPECÍFICO COORD.CLÍNICO COORD TEC ADMINISTRAT MÉDICO FARMACÊUTICO ENFERMEIRO NUTRICIONISTA SIM NÃO 7. INF Qual a periodicidade com que se reúne a EMTN? SIM NÃO 8. I Existem registros formais das reuniões da EMTN? 9. INF A UH contrata EPBS? 10. INF Qual(is) a(s) EPBS contratada(s)? A - ATIVIDADES DA EMTN? NOME: ENDEREÇO: B - FARMÁCIA: NOME: ENDEREÇO: 159
160 822 SIM NÃO 11. INF Existe(m) contrato(s) firmado(s) entre UH e a(s) EBPS especializada(s)? ATIVIDADES DA EMTN FARMÁCIA 12. INF Responsáveis na Unidade Hospitalar/EPBS: UH EPBS Diretor Clínico Diretor Técnico Coord.Tec.Adm. da EMTN Coord.Clinico da EMTN Farmacêutico Responsável Enfermeiro Responsável Nutricionista Responsável SIM NÃO 13. I Existem protocolos para os procedimentos médicos? 14. I A aplicação dos protocolos está devidamente registrada? 15. N Existem protocolos para a atuação do farmacêutico na qualidade de membro da EMTN? 15.1 INF Quais? 16. N Existem registros de sua aplicação? 17 I As atividades do enfermeiro contemplam as exigências do item C - Roteiro de Inspeção das atividades de Administração deste anexo? 18. R Existem protocolos para os procedimentos do nutricionista? 18.1 INF Quais? 19. R Existem registros de sua aplicação? SIM NÃO 20. NA EMTN oferece programa de Educação Continuada para os demais profissionais da UH/EPBS? 21. N Existem registros dos programas realizados? 22. I O Coordenador Técnico Administrativo assegura condições para o cumprimento das atribuições gerais da equipe e de seus profissionais, contido no Anexo 1? 23. N O Coordenador Técnico Administrativo representa a equipe em assuntos relacionados com as atividades da EMTN? 24. N O Coordenador Técnico Administrativo incentiva e promove programas de educação continuada para os profissionais envolvidos na TN? 25. N O Coordenador Técnico Administrativo padroniza os indicadores de qualidade para a TN? 25.1 INF Quais os indicadores de qualidade padronizados? SIM NÃO 26. I O Coordenador Clínico estabelece os protocolos de avaliação, indicação, 160
161 prescrição e acompanhamento de TN? 27. INF Com que periodicidade os protocolos são reavaliados pelo Coordenador Clínico? 28. I Os desvios de qualidade são devidamente investigados e documentados pelo Coordenador Clínico? 29. I São estabelecidas ações corretivas para os desvios de qualidade? 29.1 I Existem registros? 30. R O Coordenador Clínico assegura a atualização técnico-cientifica da EMTN? INF Como e com que frequência? 31. INF Como o Coordenador Clínico garante a qualidade dos procedimentos em relação a outros procedimentos na TN? 32. INF A estrutura da EMTN é compatível com a demanda? SIM NÃO 33. INF A EMTN é responsável por quantos leitos? 34. INF Existem outros médicos, que não da EMTN, que prescrevem TN? 35. N Existe consenso entre estes médicos e a EMTN? 36. N Existe documento comprobatório deste consenso? 37. I Existem registros das prescrições médicas? 38. R O médico orienta o paciente, familiares ou responsável legal quanto aos riscos e benefícios da TN? 39. I Existe protocolo estabelecido para utilização de cateter intravenoso central? 39.1 I Existem registros da realização deste procedimento e de suas complicações? 39.2 I Existe comprovação da localização correta do cateter intra venoso central? 40 N Existem registros da evolução médica dos pacientes submetidos à TN? SIM NÃO 41. R O nutricionista participa da evolução nutricional destes pacientes? 42. N Existem registros dos resultados de exames complementares realizados para o acompanhamento dos pacientes em TN? 43. I Existem registros da avaliação nutricional dos pacientes em TN? 43.1 INF Com que periodicidade? 44. Pessoas Contactadas durante a inspeção: 45. Conclusões 46. Local e Data 47. Nome e Número de Credencial/Assinatura dos Inspetores: B. ROTEIRO DE INSPEÇÃO PARA FARMÁCIA DE PREPARAÇÃO DE NUTRIÇÃO PARENTERAL 1. IDENTIFICAÇÃO DA FARMÁCIA a)razão SOCIAL: b)c.g.c.: / / c) NOME FANTASIA: d) ENDEREÇO: 161
162 CEP: - BAIRRO: MUNICÍPIO: UF: FONE: ( ) FAX: ( ) E.MAIL: e) Licença de funcionamento nº : afixado em local visível &127; SIM &127; NÃO f) Responsável Técnico: CRF/ n.º &127; presente &127; ausente g) FILIAL (FILIAIS) COM A MESMA ATIVIDADE: ENDEREÇO: CEP: - BAIRRO: MUNICÍPIO: UF: 824 FONE: ( ) FAX: ( ) E.MAIL: h.pessoas CONTATADAS: 2. considerações gerais SIM NÃO 2.1 R Os arredores da farmácia estão limpos e apresentam boa conservação? 2.2 R Existem fontes de poluição ou contaminação ambiental próximas à farmácia? 2.3 N Existe proteção contra a entrada de roedores, insetos, aves ou outros animais? 2.4 N Existe programa de sanitização, desratização e desinsetização? INF Qual a periodicidade? N Existem registros? 2.5 N Os esgotos e encanamentos estão em bom estado? 2.6 INF Nº total de funcionários: (M) (F) Nível superior: Outros: 2.7 N As atribuições e responsabilidades estão formalmente descritas e entendidas pelos envolvidos? 2.8 N Os funcionários são submetidos a exames médicos periódicos? INF Qual a Periodicidade? N Existem registros? 2.9 R Existem sanitários em quantidade suficiente? R Estão limpos? 2.10 R Existem vestiários em quantidade suficiente? R Estão limpos? 2.11 INF Existe local para refeições? INF Se não, onde os funcionários fazem suas refeições? 2.12 R Os funcionários estão uniformizados? R Os uniformes estão limpos e em boas condições? 162
163 2.13 N São realizados treinamentos dos funcionários? N Existem registros? 2.14 N Existe gerador próprio ou outro sistema para o caso de falta de energia elétrica? INF Qual o procedimento adotado? 2.15 Observações 3. ÁREA DE DISPENSAÇÃO SIM NÃO 3.1 INF Qual a área ocupada pelo setor em m2? 3.2. R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 3.3. R As paredes são de cor clara, lisas e estão em bom estado de conservação? 3.4. R O teto está em boas condições? 3.5 R O setor está limpo? 3.6. R A iluminação é suficiente e adequada? 3.7 R A ventilação do local é suficiente e adequada? 3.8 I A manipulação da N P é feita somente sob prescrição médica? INF Quais os mecanismos de recebimento das prescrições? 3.9 INF Existe um sistema de Registro Geral das prescrições médicas? INF Qual? I Todas as prescrições estão devidamente registradas? 3.10 Observações: ÁREA DE ARMAZENAMENTO SIM NÃO 4.1. R A área tem capacidade suficiente para assegurar a estocagem ordenada e racional das diversas categorias de materiais, produtos farmacêuticos e correlatos? 4.2. N O local oferece condições de temperatura adequada para o armazenamento de materiais, produtos farmacêuticos e correlatos? R Existe controle de temperatura e umidade? R Existem registros? 4.3 R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 4.4 R As paredes estão bem conservadas? 4.5 R O teto está em boas condições? 4.6. R O setor está limpo? 4.7 R A qualidade e a intensidade da iluminação são adequadas? 4.8 R A ventilação do local é suficiente e adequada? 4.9 R As instalações elétricas estão em bom estado de conservação, segurança e uso? 4.10 N Existem equipamentos de segurança para combater incêndios? N Os extintores estão dentro do prazo de validade? N O acesso aos extintores e mangueiras está livre? 4.11 INF Existe necessidade de câmara frigorífica e ou geladeira? R A câmara frigorífica e ou geladeira é mantida limpa sem acúmulo de gelo? N Existe controle e registro de temperatura? 163
164 4.12 R Os produtos farmacêuticos, correlatos e materiais de embalagem estão armazenados afastados do piso e paredes, facilitando a limpeza? N Existe local segregado para estocagem de produtos farmacêuticos, correlatos e materiais de embalagem reprovados, recolhidos ou para devolução? R Existem recipientes para lixo com tampa e estão devidamente identificados? N As aberturas e janelas encontram -se protegidas contra a entrada de aves, insetos, roedores e outros animais? 4.16 N Os produtos farmacêuticos, correlatos, e materiais de embalagem são inspecionados quando do seu recebimento? N Os produtos farmacêuticos, correlatos e materiais de embalagem estão devidamente identificados? I Os produtos farmacêuticos e correlatos, possuem registros no Ministério da Saúde e estão dentro do prazo de validade? I Os produtos farmacêuticos e correlatos são acompanhados dos respectivos laudos de análises dos fornecedores, devidamente assinados pelos seus responsáveis? 4.17 R Existe sistema de controle de estoque? &127; fichas &127; informatizado SIM NÃO 4.18 R O uso dos produtos farmacêuticos e correlatos respeita a ordem utilizando-se primeiro o mais antigo? 4.19 N Os produtos farmacêuticos e correlatos que não são aprovados na inspeção de recebimento (item 4.16) são rejeitados e devolvidos ou destruídos? N Existem registros? 4.20 R Existem procedimentos operacionais escritos para as atividades do setor? 4.21 Observações: 5. ÁGUA SIM NÃO 5.1 N As instalações de água potável atendem às exigências deste Regulamento? N É procedida limpeza da caixa d'água? INF Qual a periodicidade? R Existem procedimentos escritos para limpeza do depósito de água potável? N Existem registros das limpezas efetuadas? 5.3 INF A água potável é submetida a algum processo de purificação? INF Qual? SIM NÃO N São realizados controles microbiológicos da água potável? INF Qual a periodicidade? 5.4 INF Para que se destina a água? &127; limpeza de material &127; preparação de álcool a 70% &127; fabricação de produtos farmacêuticos SIM NÃO 5.5 I Existem registros que comprovem as especificações fisico-químicos e 164
165 microbiológicos da água para utilizada? 5.6 INF A água para injetáveis utilizada é industrializada? 5.7 Observações: ÁREAS DE PREPARAÇÃO SIM NÃO 6.1 INF As áreas destinadas à preparação da NP são adequadas e suficientes ao desenvolvimentos das operações, dispondo de todos os equipamentos de forma organizada e racional? 6.2 INF A Circulação de pessoal nestas áreas é restrita? 6.3 R É proibida a entrada de pessoas não autorizadas nos diversos setores da área de preparação? 6.4 I A área destinada à preparação da N P possui: &127; área de limpeza e higienização de produtos farmacêuticos e correlatos? &127; vestiário (antecâmara)? &127; área de manipulação? &127; área de rotulagem/embalagem? SIM NÃO 6.5 N Existem equipamentos de segurança para combater incêndios, atendendo à legislação específica? 6.6 Observações: 7. ÁREA DE LIMPEZA E HIGIENIZAÇÃO SIM NÃO 7.1. I Existe local próprio para limpeza e higienização de materiais, produtos farmacêuticos e correlatos? I Está localizado anexo à área de manipulação? INF Qual a classificação desta área? SIM NÃO 7.2 N O piso é liso, resistente e de fácil limpeza? N O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 7.3 N As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 7.4 N As janelas e ou visores existentes nos diversos setores da área de preparação estão perfeitamente vedados? 7.5 N A iluminação é suficiente e adequada? 7.6 N A ventilação é suficiente e adequada? N Existem equipamentos de segurança para combater incêndio atendendo à legislação específica? N O acesso aos extintores e mangueiras está livre? N Os extintores estão dentro do prazo de validade? 7.8 I Existe passagem de dupla porta para entrada de produtos farmacêuticos, correlatos e materiais na área de manipulação? 7.9 INF Existem ralos? N São sifonados? 165
166 7.10 N Dispõe de meios e equipamentos adequados para limpeza prévia das embalagens dos produtos farmacêuticos e correlatos? 7.11 N Os produtos utilizados para assepsia dos produtos farmacêuticos, correlatos e materiais de embalagem obedecem às especificações doministério da Saúde? 7.12 R Existem procedimentos escritos para higienização de produtos farmacêuticos, correlatos e materiais de embalagem? 7.13 N Os procedimentos de higienização garantem a assepsia e mantêm a qualidade dos produtos farmacêuticos, correlatos e materiais de embalagem? 7.14 N Existe sistema de inspeção visual para revisão dos produtos farmacêuticos e correlatos? 7.15 N A transferência dos produtos farmacêuticos, correlatos e materiais de embalagem para a área de manipulação da NP se realiza em condições de segurança, atendendo às especificações desta norma? 7.16 R Existe recipiente para lixo? 7.17 Observações: 8. VESTIÁRIO (ANTECÂMARA) SIM NÃO 8.1 INF As áreas destinadas a vestiário são adequadas e suficientes ao desenvolvimento das operações, dispondo de todos os equipamentos de forma organizada e racional? 8.2 N O piso é liso, resistente e de fácil limpeza? N O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 8.3 N As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 8.4 N As janelas e ou visores existentes nos diversos setores da área de preparação estão perfeitamente vedados? 8.5 N A iluminação é suficiente e adequada? 8.6 R Existe sistema de travas e de alerta visual ou auditivo para controlar o acesso ao vestiário (antecâmara) à área de manipulação? 8.7 N Existe sistema de filtração de ar? 8.8 N A pressão do ar no vestiário (antecâmara) é inferior à da sala de manipulação da NP, mas superior à das outras dependências? 8.9 INF Equipamentos Existentes: a. pia e torneira: &127; sem pedal &127; com pedal &127; com alavanca para cotovelo &127; com célula foto elétrica b. &127; dispensadores para degermantes ou antissépticos c. &127; toalhas descartáveis d. &127; secador a ar e. &127; armários para guardar uniformes limpos/esterilizados 828 f. &127; cesto para despejo de roupas usadas 166
167 g. &127; outros. Especificar: 8.10 INF Quais os produtos utilizados para degermação das mãos? SIM NÃO 8.11 R Existem procedimentos escritos para a paramentação e higienização das mãos: 8.12 Observações 9. ÁREA DE MANIPULAÇÃO 9.1 INF Qual a área ocupada pelo setor em m2? 9.2 INF Qual o n.º de funcionários que atuam na área, por turno? INF Qual a formação profissional dos funcionários? SIM NÃO 9.3 N O piso é liso, resistente e de fácil limpeza? O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 9.4 N As paredes e teto são de cor clara, lisas, impermeáveis e resistentes aos agentes sanitizantes e possuem cantos arredondados? 9.5 INF Existem ralos? 9.6 INF O local é utilizado para manipulação e ou fracionamento de outras preparações? INF Quais? SIM NÃO 9.7 I O manipulador confere cuidadosamente a identificação do paciente e sua correspondência com a formulação prescrita antes e após a sua manipulação? 9.8 I A área possui pressão positiva? INF Qual a classificação desta área? SIM NÃO 9.9 I Existe controle sistemático do nível de contaminação do ar? INF Qual a frequência? SIM NÃO I Existem registros? 9.10 I Existe equipamento de fluxo laminar? 9.11 I O ar injetado na área é filtrado? INF Qual o tipo de filtro? SIM NÃO 9.12 N O ar injetado no fluxo laminar é filtrado por filtros HEPA? 9.13 N É verificado o estado dos filtros de ar de ingresso á área e do equipamento de fluxo laminar? INF Qual a frequência? SIM NÃO N Existem registros? 9.14 N São realizados controles para determinar a contagem de partículas? I Qual a frequência? SIM NÃO N -Existem registros? 9.15 I O fluxo laminar está validado? I Existem registros? 167
168 9.16 I São feitos controles microbiológicos do ar e das superfícies e de pessoal? INF Qual a frequência? SIM NÃO I Existem registros? 9.17 I Os manipuladores estão devidamente uniformizados? I Os uniformes são confeccionados de tecido que não liberam partículas? I Os uniformes são esterilizados? INF Qual a frequência de troca dos uniformes? 9.18 INF Qual a frequência de troca das luvas estéreis durante o trabalho de manipulação? SIM NÃO 9.19 N Existem procedimentos escritos para garantir que a entrada de produtos farmacêuticos e correlatos na área de preparação seja realizada de forma segura? SIM NÃO 9.20 N Existem procedimentos escritos para limpeza da área? N Existem registros? 9.21 N Existe procedimento escrito para limpeza do fluxo laminar? N Existe registro? 9.22 N Existem registros do número de lote de cada um dos produtos farmacêuticos e dos correlatos utilizados na manipulação de cada prescrição de NP indicando inclusive os seus fabricantes? 9.23 I Os recipiente utilizados para acondicionamento da NP atendem às especificações deste Regulamento? 9.24 INF Qual a frequência da troca do equipo de transferência de produtos farmacêuticos? SIM NÃO 9.25 N Existem procedimentos escritos que garantam a saída da NP para a área de embalagem de maneira segura? 9.26 INF Como ocorre a saída da NP preparada para a área de embalagem? 9.27 INF Qual o destino das sobras dos componentes utilizados na manipulação da NP? 9.28 INF Como é procedido o descarte do material utilizado na manipulação da NP? SIM NÃO 9.29 N As condições da área são condizentes com o volume das operações realizadas por turno de trabalho? 9.30 Observações: 10. ÁREA DE EMBALAGEM SIM NÃO R Existe área própria para embalagem? 10.2 R O piso é liso, resistente e de fácil limpeza? R O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 10.3 R As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 168
169 10.4 R A iluminação é suficiente e adequada? 10.5 R A ventilação é suficiente e adequada? 10.6 INF Quais os equipamentos existentes? SIM NÃO 10.7 I São realizados controles para verificar se a NP foi preparada conforme prescrição médica? INF Quais os controles realizados? SIM NÃO 10.8 I Os rótulos apresentam todas as informações exigidas por este Regulamento? 10.9 N O acondicionamento da NP já rotulado atende às especificações deste Regulamento? Observações CONSERVAÇÃO E TRANSPORTE SIM NÃO 11.1 I Existe refrigerador, exclusivo para medicamentos, com termômetro para conservação da NP até o momento do seu transporte? N Existem registros do controle sistemático da temperatura? 11.2 INF Como é realizado o transporte da NP? SIM NÃO 11.3 I Os recipientes térmicos utilizados para o transporte da NP garantem a manutenção interna da temperatura dentro da faixa pré estabelecida? 11.4 I As condições de acondicionamento para o transporte da NP estão validadas? I Existem registros? 11.5 I A NP durante o transporte se mantém protegida das intempéries e da incidência direta da luz solar? 11.6 N Existem procedimentos operacionais escritos? 11.7 Observações 12. GARANTIA DA QUALIDADE 12.1 N A farmácia possui um sistema de Garantia da Qualidade implantado, com base nas diretrizes das BPPNP? 12.2 I Existem procedimentos operacionais normatizados para todas as operações críticas da preparação e de controle de qualidade da NP? SIM NÃO 12.3 N São realizadas auto-inspeções? INF Com que frequência? SIM NÃO N Existem registros? 12.4 N Existe um programa de treinamento para todos os funcionários? N Existem registros? 12.5 N A documentação referente à preparação da NP são arquivadas ordenadamente durante 5 anos? 12.6 N A documentação existente possibilita o rastreamento para investigação de 169
170 qualquer suspeita de desvio de qualidade da NP? 12.7 N Existem registros de reclamações referentes a desvios de qualidade de NP? N Existem registros das investigações e correções, bem como das ações corretivas? INF As conclusões das investigações são transmitidas por escrito ao reclamante? 12.8 Observações 13. CONTROLE DE QUALIDADE SIM NÃO 13.1 R Existe laboratório de Controle de Qualidade no estabelecimento? 13.2 INF A empresa realiza ensaios específicos com terceiros? INF Quais? INF Com quem? SIM NÃO 13.3 I O Controle de Qualidade possui pessoal técnico qualificado para exercer a função? 13.4 N O piso é liso, resistente e de fácil limpeza? N O estado de higiene e conservação do piso é bom, sem buracos e rachaduras? 13.5 N As paredes e o teto são de cor clara, lisas e estão em bom estado de conservação? 13.6 INF Existem ralos? N São sifonados? 13.7 N As instalações elétricas, de água e de gás estão em boas condições de uso? 13.8 N A iluminação é suficiente e adequada? SIM NÃO 13.9 N A ventilação é suficiente e adequada? N Existem equipamentos de segurança para combater incêndios? N Os extintores estão dentro do prazo de validade? N O acesso aos extintores e mangueiras está livre? N Existem equipamentos de proteção e segurança individual (ducha, lava-olhos, óculos, etc.)? R A área de circulação está livre de obstáculos? R Existem registros? N Existe programa de limpeza e manutenção periódica de equipamentos e aparelhos? N Existem especificações escritas para a aquisição dos produtos farmacêuticos, correlatos e recipientes? N A especificação exige o fornecimento do certificado de análise dos produtos farmacêuticos, correlatos e recipientes? N O Controle de Qualidade monitora o cumprimento dos procedimentos de limpeza, higienização e sanitização da preparação da NP? N Existem métodos analíticos utilizados para a realização das análises? N São realizadas análises nas NPs preparadas conforme estabelecido neste Regulamento? 170
171 INF Em caso negativo, qual a metodologia e critério de amostragem adotado? SIM NÃO N Existem registros? I Amostras de contra-prova de cada NP manipulada são conservadas sob refrigeração à temperatura de 20 C a 80 C durante 7 dias após o seu prazo de validade? N Existem procedimentos operacionais escritos para o setor? Observações 14. CONCLUSÃO 15. NOME, Nº DE CREDENCIAL E ASSINATURA DOS INSPETORES 16. DATA C ROTEIRO DE INSPEÇÃO PARA ATIVIDADES DE ADMINISTRAÇÃO DE NUTRIÇÃO PARENTERAL 1 - IDENTIFICAÇÃO DO LOCAL DAS ATIVIDADES DE ADMINISTRAÇÃO DA NUTRIÇÃO PARENTERAL ( ) HOSPITAL Setor UTI Clínica Cirúrgica Pediatria Clínica Médica n de leitos n.º de enfermeiros n de técnicos de enfermagem n de auxiliares de enfermagem ( ) AMBULATÓRIO ( ) RESIDÊNCIA Endereço: cep: Bairro: Município: uf: fone: ( ) RESPONSÁVEL TÉCNICO: COREN/ Nº &127; PRESENTE &127; AUSENTE 2. CONSIDERAÇÕES GERAIS SIM NÃO 2.1. I A NP é administrada sob a responsabilidade do Enfermeiro? 2.2 INF Se não é administrada por Enfermeiro, indique quem administra: 2.3 I Tem Enfermeiro de plantão quando da administração da NP? INF Em período: PARCIAL TOTAL SIM NÃO 2.4. N Existe disponibilidade do Enfermeiro Responsável pelo atendimento ao paciente em NP domiciliar? INF De que forma? &127; VISITAS &127; TELEFONE &127; BIP 171
172 SIM NÃO 2.5 N O Enfermeiro participa do processo de seleção, padronização, licitação e aquisição de equipamentos e materiais para a administração e controle da NP? 2.6 N Há treinamento inicial e contínuo voltado para a administração da NP e utilização de bombas infusoras? INF Qual a periodicidade do treinamento? SIM NÃO R O treinamento segue uma programação preestabelecida? R Há registros do treinamento? 2.7 N Existe manual de procedimentos para a administração da NP atualizado? N O manual de procedimentos está disponível para consulta imediata por todos os funcionários? 2.8 N Na admissão dos funcionários são realizados exames médicos e laboratoriais? INF Esses exames são repetidos com que periodicidade? SIM NÃO N Existem registros desses exames? 2.9 N Os funcionários estão utilizando uniformes próprios para a atividade? N Os uniformes estão limpos e em boas condições? 2.10 INF Há lavatórios em número suficiente? N Existe sabão, papel toalha ou aparelho de ar para secagem das mãos disponíveis e em quantidade suficiente? R Existe folheto ilustrativo ou recomendação para lavagem das mãos próximo às pias? 2.11 N Os funcionários não usam jóias ou relógios? 2.12 N Os funcionários usam gorro e máscara no manuseio da NP? SIM NÃO 2.13 R São utilizadas bombas infusoras na administração da NP? R As bombas infusoras são adequadas à faixa etária dos pacientes? N Existe procedimento escrito de limpeza e desinfecção das bombas infusoras? N Há registros dessas operações? N As bombas infusoras apresentam etiqueta indicando as datas da última e da próxima calibração? R Existe um programa por escrito de manutenção das bombas infusoras de forma: &127; PREVENTIVA &127; CORRETIVA SIM NÃO N As bombas infusoras são submetidas à manutenção periódica? INF Quem realiza a manutenção das bombas infusoras? &127; HOSPITAL &127; FORNECEDOR &127; TERCEIRIZADO 172
173 SIM NÃO N Há registros da manutenção? N Existem procedimentos escritos da operacionalização das bombas infusoras? Há fornecimento constante e em número suficiente de equipos adequados para as bombas infusoras? 2.14 R É realizada orientação ao paciente e/ou família? INF A orientação é realizada de forma: &127; VERBAL &127; ESCRITA 2.15 INF Local de realização do acesso intravenoso central? &127; CENTRO CIRÚRGICO &127; ENFERMARIA &127; UTI &127; OUTRO. QUAL? SIM NÃO 2.16 I Existe material de reanimação para caso de emergência? N O material encontra-se em local de fácil acesso? I O material está limpo e em condições de uso? 2.17 N Existe procedimento escrito para realização e troca do curativo do local do acesso intravenoso de localização central? 2.18 R A unidade de radiologia é de fácil acesso? 2.19 R Existe horário estabelecido para a entrega das prescrições na Farmácia? 2.20 I Quando do recebimento da NP da Farmácia, são observados: &127; INTEGRIDADE DA EMBALAGEM &127; PRESENÇA DE PARTÍCULAS NA NP &127; NOME DO PACIENTE/Nº DO LEITO &127; COMPOSIÇÃO E VOLUME TOTAL DA NP &127; PRAZO DE VALIDADE DA NP &127; RECOMENDAÇÕES ESPECÍFICAS &127; OUTRO. QUAL? INF Quando observada qualquer anormalidade, no recebimento da NP, qual o procedimento adotado? SIM NÃO 2.21 I Quando não usada imediatamente, a NP é conservada em geladeira exclusiva para medicamentos? I Existe controle e registro sistemático de temperatura da geladeira? N A geladeira encontra-se limpa, sem acúmulo de gelo e em boas condições de 834 conservação? R Existe procedimento escrito de limpeza e desinfecção da geladeira? 2.22 N O local de manuseio da NP está em boas condições de conservação, organização 173
174 e limpeza? N Há procedimento escrito para limpeza e desinfecção da área e do local de manuseio da NP? 2.23 I Quando da administração da NP, são observados: &127; INTEGRIDADE DA EMBALAGEM &127; PRESENÇA DE PARTÍCULAS NA NP &127; NOME DO PACIENTE/Nº DO LEITO &127; COMPOSIÇÃO E VOLUME TOTAL DA NP &127; PRAZO DE VALIDADE DA NP &127; RECOMENDAÇÕES ESPECÍFICAS &127; OUTRO. QUAL? SIM NÃO 2.24 N A NP está protegida da incidência direta de luz? 2.25 N A NP é protegida das fontes geradoras de calor durante a sua administração? 2.26 N A via de acesso é exclusiva para administração da NP? INF Em casos excepcionais, a autorização para utilização da via de acesso da NP é: &127; VERBAL &127; ESCRITA SIM NÃO 2.27 R São utilizados filtros na linha de infusão da NP? 2.28 I A NP é administrada diretamente do seu recipiente de origem? 2.29 I É realizada antissepsia nas conexões do catéter na troca do equipo? INF A antissepsia é realizada com que solução? 2.30 INF No caso de descontinuidade da infusão da NP, como é mantida a via de acesso? SIM NÃO 2.31 N Há registros de todo o processo de administração da NP? 2.32 I É realizado o controle clínico e laboratorial no paciente em NP? INF Quais? &127; PESO &127; SINAIS VITAIS &127; PRESSÃO ARTERIAL &127; GLICEMIA CAPILAR &127; GLICOSÚRIA &127; BALANÇO HÍDRICO SIM NÃO 2.33 N Os exames clínicos e laboratoriais são realizados em tempo hábil? 2.34 N Há registros de todo o processo de administração da NP? INF Quais os impressos utilizados? 174
175 &127; FICHA DE EVOLUÇÃO DE ENFERMAGEM &127; LIVRO DE RELATÓRIO DE ENFERMAGEM &127; FICHA DE BALANÇO HÍDRICO 835 &127; OUTROS. QUAIS? SIM NÃO 2.35 I Há registros dos exames clínicos e laboratoriais? 2.36 N É realizada avaliação do paciente antes da interrupção/suspensão da TN? 2.37 N Há registros da avaliação realizada? 3. PESSOAS CONTACTADAS 4. OBSERVAÇÕES 5. CONCLUSÕES 6. NOME, Nº DE CREDENCIAL E ASSINATURA DOS INSPETORES 7. DATA ANEXO VI INFORME CADASTRAL DE UH OU EPBS PARA A PRÁTICA DA TN A - IDENTIFICAÇÃO DA UH/EPBS A1 - IDENTIFICAÇÃO DA EMPRESA a) RAZÃO SOCIAL: b) C.G.C.: / / c) NOME FANTASIA: d) ENDEREÇO: CEP: - BAIRRO: MUNICÍPIO: UF: FONE: ( ) FAX: ( ) E.MAIL: e) TIPO DE EMPRESA UNIDADE HOSPITALAR (UH) EMPRESA PRESTADORA DE BENS E SERVIÇOS (EPBS) A2 ATIVIDADES DA UH/EPBS 1 A UH/EPBS deseja cadastrar-se como provedora de atividades inerentes à TN desempenhadas por SIM NÃO 1.1 Preparação de NP? 1.2 Indicação, prescrição, administração, controle clinico laboratorial e avaliação final? 1.3 Todas as atividades. (Preencher todo o informe) 175
176 2. A EMTN foi constituída por ato formal em de de, segundo o documento (xerox anexo) 3. A composição da EMTN compreende: Nome RG CIC N.Inscr. Conselho Membros COORDENADOR CLINICO COORDENADOR TéCNICO ADM. Médico FARMACÊUTICO 836 ENFERMEIRO NUTRICIONISTA OUTROS, ESPECIFICAR 4. Os membros da EMTN possuem os seguintes títulos de especialista ou de habilitação devidamente documentados e registrados em conselhos ou associações de classe : 4.1. MEMBROS NÃO TÍTULO ESPECIALISTA Expedido pela Sociedade Ano ou HABILITAÇÃO. Ano Brasileira de: COORD TEC ADM. COORD CLINICO. MEDICO FARMACÊUTICO ENFERMEIRA NUTRICIONISTA 4.2. MEMBROS MESTRADO (ANO) LIVRE DOCÊNCIA ANO Coord.Tec.Adm. da EMTN DOUTORADO (ano) Coord.Clinico da EMTN Farmacêutico Responsável Enfermeiro Responsável Nutricionista Responsável 5. A EMTN possui protocolos para os procedimentos multiprofissionais de:? Médicos Enfermagem Farmácia Nutricionista 6. Formação profissional na área de TN dos componentes da EMTN, comprovadas por documentos apresentados Membros Residência Estágios Cursos Congressos Coord. Clin Coord Tec Ad Médico Farmacêutico Enfermeiro Nutricionista 176
177 SIM NÃO 7. A EMTN dispõe de programa de Educação Continuada para os demais profissionais da UH/EPBS? 8. A EMTN dispõe comprovadamente de : indicadores de qualidade para a TN? SIM NÃO protocolos de avaliação, indicação, prescrição e acompanhamento SIM NÃO programas de educação continuada para os profissionais envolvidos na TN? SIM NÃO metodologia para investigar e registrar desvios de qualidade? SIM NÃO A ETN está preparada para assegurar sua atualização técnico-cientifica?sim NÃO 9. Existe protocolo estabelecido para realização de acesso intravenoso central? SIM NÃO 10. Existem formulários para registros da : avaliação nutricional dos pacientes em TN? SIM NÃO evolução médica diária dos pacientes submetidos a TN? SIM NÃO 837 resultados de exames complementares para o acompanhamento datn? SIM NÃO 11. Conclusão: A empresa (não) está cadastrada e em condições de desempenhar atividades de Farmácia EMTN em terapia nutricional 12. Local e Data 13. Nome e Número de Credencial/Assinatura dos Inspetores. 177
178 PORTARIA Nº 40, DE 13 DE JANEIRO DE 1998 Regulamento que estabelece normas para Níveis de Dosagens Diárias de Vitaminas e Minerais em Medicamentos ORIGEM: Grupo de Trabalho instituído pela Portaria SVS/MS n 254, publicada no D.O.U. de 24 de junho de Considerando: a) a necessidade de fixar níveis para a recomendação diária de consumo de vitaminas e minerais em medicamentos; b) a necessidade de estabelecer diretrizes claras aos fabricantes para a formulação e recomendação posológica destas substâncias em medicamentos; c) a necessidade de estabelecer regras bem definidas que permitam diferenciar claramente o que sejam "Medicamentos à Base de Vitaminas e ou Minerais ou suas Associações" (definidos no âmbito da Lei n 6360 de 23 de setembro de 1976, regulamentada pelo Decreto n de 5 de janeiro de 1977) dos "Suplementos Vitamínicos e ou Minerais", definidos no âmbito do Decreto-Lei n 986 de 21 de outubro de 1969; d) a necessidade de regulamentar a importação de produtos submetidos ao regime de vigilância sanitária; e) a Portaria SNVS/MS nº 64, de 28 de dezembro de 1984; f) que as Resoluções Normativas n s 2 e 3/78, da Câmara Técnica de Medicamentos, não mais atendem ao estágio atual do conhecimento; g) os estudos sobre níveis seguros de vitaminas e minerais realizados pelo Grupo de Trabalho designado pela Portaria n 254, de 24 de junho de 1997; h) a necessidade de constante aperfeiçoamento das ações de controle sanitário visando a proteção da qualidade a que deverão obedecer os MEDICAMENTOS À BASE DE VITAMINAS E MINERAIS; Resolve: Art.1º Definir, sem prejuízo do disposto na Lei n 6.360, de 23 de setembro de 1976 e no seu regulamento, o Decreto n , de 5 de janeiro de 1977, como "Medicamentos à base de vitamina isolada, vitaminas associadas entre si, minerais isolados, minerais associados entre si e de associações de vitaminas com minerais", aqueles cujos esquemas posológicos diários situam-se acima dos 100% da Ingestão Diária Recomendada - IDR (estabelecida por legislação específica) de acordo com os níveis definidos nesta Portaria. Art.2º Consideram-se os medicamentos definidos no artigo anterior, como de "Venda Sem Exigência de Prescrição Médica" quando os níveis diários indicados para quaisquer dos componentes ativos, objeto deste regulamento, situem-se até os limites considerados seguros, constantes da tabela anexa. Art.3º Consideram-se os medicamentos definidos no artigo 1, como de "Venda Com Exigência de Prescrição Médica", quando os níveis diários indicados dos componentes ativos situem-se acima dos limites considerados seguros por este regulamento, ou sempre que estiverem contidos em formulações para uso injetável. Art. 4º No caso de associações entre as substâncias, objeto desta norma, a presença na formulação de pelo menos um componente nas faixas de dose previstas no artigo 3º deste regulamento, já enquadra o produto nas condições previstas no respectivo artigo. Art.5º Para melhor informar o consumidor, deve constar na embalagem dos medicamentos nacionais ou importados, objeto desta Portaria, a formulação qualitativa e quantitativa por unidade farmacotécnica e o teor percentual do(s) componente(s) na dose/posologia diária máxima preconizada, expresso claramente em índices percentuais, relativos à IDR. 178
179 Art.6º O registro dos produtos referidos neste regulamento está sujeito às exigências gerais para Registro e Rotulagem de Medicamentos, previstos na legislação. Art.7º Para fins desta Portaria, consideram-se Níveis Seguros de Vitaminas e ou Minerais para as doses diárias indicadas em Medicamentos, aqueles constantes na tabela anexa. Art. 8º As empresas tem o prazo de 180 (cento e oitenta) dias, a contar da data de publicação deste Regulamento, para se adequar ao mesmo. Art. 9º Ficam revogadas as Resoluções Normativas n 2/78, da Câmara Técnica de Medicamentos, do Conselho Nacional de Saúde, de 6 de novembro de 1978, e n 3/78 da Câmara Técnica de Medicamentos, do Conselho Nacional de Saúde, de 3 de outubro de 1978 e demais disposições em contrário. MARTA NOBREGA MARTINEZ ANEXO - NÍVEIS MÁXIMOS DE SEGURANÇA DE VITAMINAS E OU MINERAIS Componente Dose Diária para Adultos Dose Diária para Pediatria Vitamina A* UI - Lactentes 500 UI/kg peso corporal até o limite de 5000 UI - Pediátrico 500 UI/kg até o limite de UI Beta-caroteno** 25 mg - Lactentes 500 UI/kg peso corporal até o limite de 5000 UI - Pediátrico 500 UI/kg até o limite de UI Vitamina D 800 UI - Lactentes 40 UI/kg peso corp. até o limite de 400 UI - Pediátrico 40 UI/kg até o limite de 800 UI Vitamina E UI Lactentes 20 UI/kg peso corporal até o limite de 200 UI Pediátrico 20 UI/kg peso corporal até o limite de 400 UI Vitamina C mg Lactentes 25 mg/kg peso corp. até o limite de 300 mg Pediátrico 25 mg/kg até o limite de 1000 mg Vitamina B6-Piridoxina 200 mg -Lactentes 10 mg/kg peso 179
180 corp. até o limite de 100 mg -Pediátrico 10 mg/kg peso corporal até o limite de 200 mg Vitamina B2-Riboflavina 200 mg -Lactentes 10 mg/kg peso corp. até o limite de 100 mg -Pediátrico 10 mg/kg peso corporal até o limite de 200 mg Vitamina B5 ou PP ou Niacina (sob a forma de Niacinamida, não se recomendando sob a forma ácida) 500 mg -Lactentes 20 mg/kg peso corp. até o limite de 200 mg -Pediátrico 20 mg/kg peso corporal até o limite de 400mg Vitamina B1-Tiamina 200 mg -Lactentes 10 mg/kg peso corp. até o limite de 100 mg -Pediátrico 10 mg/kg até o limite de 200 mg Vitamina B12 Cobalaminaas mcg -Lactentes 50 mcg/kg peso corp. até o limite de 500 mcg -Pediátrico 50 mcg/kg até o limite de 1000 mcg Ácido Fólico 1 mg -Lactentes 10 mcg/kg peso corp. até o limite de 100 mcg -Pediátrico 10 mcg/kg até o limite de 300 mcg Vitamina K 25 mg -Lactentes 1 mg/kg peso corp. até o limite de 10 mg -Pediátrico 1 mg/kg até o limite de 25mg Ácido Pantotênico mg -Lactentes 50mg/kg peso corp. até o limite de 500 mg -Pediátrico 50 mg/kg até o limite de 1000 mg Biotina 2,5 mg -Lactentes 0,125 mg/kg peso corp. até o limite de 1,25 mg -Pediátrico 0,125mg/kg até o limite de 2,5 mg Cálcio mg -Lactentes 150 mg/kg peso corporal até o limite
181 mg -Pediátrico 80 mg/kg peso corporal até o limite de 1500 mg Fósforo mg -Lactentes 150 mg/kg peso corporal até o limite 1200 mg -Pediátrico 80 mg/kg peso corporal até o limite de 1500 mg Magnésio 700 mg -Lactentes 10 mg/kg peso corporal até o limite 80 mg -Pediátrico 10 mg/kg peso corporal até o limite de 200 mg Ferro - Obs.: Produtos contendo Ferro elementar devem obrigatoriamente estar contidos em acondicionamentos com dispositivo de segurança para evitar ingestão indevida. Fluor 65 mg -Lactentes 2,0 mg/kg peso corporal até o limite 15 mg -Pediátrico 2,0mg/kg peso corporal até o limite de 50 mg 4,0 mg - Obs.: Este limite é - Lactentes 0,1 mg/kg de mantido apenas para peso corporal até o limite respeitar a IDR adotada. No 0,5 mg -Pediátrico 0,1 entanto, em função do mg/kg de peso corporal até potencial tóxico, recomendase 2,9 o limite de 2,0 mg mg Zinco 30 mg -Lactentes e Pediátrico 0,5 mg/kg de peso corporal até o limite 10 mg Cobre 9 mg -Lactentes 0,1 mg/kg de peso corporal até o limite 1 mg -Pediátrico 0,1 mg/kg de peso corporal até o limite de 2 mg Manganês 10 mg -Lactentes 0,1 mg/kg de peso corporal até o limite 1 mg -Pediátrico 0,1 mg/kg de peso corporal até o limite de 3 mg Molibdênio 350 mcg -Lactentes 15 mcg/kg de peso corporal até o limite 150 mcg -Pediátrico 15 mcg/kg de peso corporal até o limite de 300 mcg 181
182 Selênio 150 mcg -Lactentes 5 mcg/kg de peso corporal até o limite 50 mcg -Pediátrico 5 mcg/kg de peso corporal até o limite de 100 mcg Cromo mcg -Lactentes 10 mcg/kg de peso corporal até o limite de 100 mcg -Pediátrico 10 mcg até o limite de 500 mcg Iodo 600 mcg -Lactentes 10 mcg/kg de peso corporal até o limite de 100 mcg -Pediátrico 10 mcg/kg de peso corporal até o limite de 300 mcg * O total de UI de Vitamina A mencionado pode ser proveniente de Retinol Equivalente e de Betacaroteno em formulações em que estiverem associados. ** Quando se tratar de única fonte de Vitamina A no medicamento NOTA: Estanho, Boro, Níquel, Silício, Vanádio: a utilidade e validade da inclusão destes componentes em medicamentos não estão claramente estabelecidas. A sua inclusão fica condicionada à apresentação de trabalhos farmacológicos e clínicos que justifiquem sua presença qualitativa e quantitativa. REFERÊNCIAS: a) Rapport sur les Limites de Sécurité dans les Consommations Alimentaires des Vitamines et Minéraux, Ministère de l'économie et des Finances Ministère du Travail et Affaires sociales Ministère de lágriculture, de la Pêche et de l'alimentation b) Vitamin and Mineral Safety, Council for Responsible Nutrition, Washington,
183 PORTARIA Nº 108, DE 25 DE JULHO DE 1991 Normaliza a composição de produtos para TRO, de acordo com os conceitos de reidratação, manutenção e prevenção em TRO contidos nas normas de controle de doenças diarreicas do Ministério da Saúde. O CHEFE DA DIVISÃO DE PRODUTOS, do Departamento Técnico-Normativo, da Secretaria Nacional de Vigilância Sanitária, do Ministério da Saúde, no uso da competência que lhe foi delegada pelo Ministro de Estado da Saúde, no termos da Portaria nº 767, de 12 de junho de 1990, e em cumprimento a dispositivos da Lei nº 6.360/76, do Decreto nº /77 e Decreto-Lei nº 986/69, e considerando; - que a doença diarreica aguda se constitui em uma das principais causas de mortalidade infantil no país; - que a terapia de reidratação oral (TRO) em um contexto de assistência materno-infantil, pode salvar a vida de milhares de crianças no país; - que os princípios básicos da TRO já estão bem fundamentados pela OMS e pela Comunidade Científica Nacional e Internacional; - que há necessidade de adequar a esses princípios as fórmulas de produtos para hidratação oral atualmente existentes no Brasil, ou aquelas que venham a ser registradas futuramente, resolve: 1. Normalizar a composição de produtos para TRO, de acordo com os conceitos de reidratação, manutenção e prevenção em TRO contidos nas normas de controle de doenças diarreicas do Ministério da Saúde. 2. Determinar o conteúdo mínimo de informações que devem ser fornecidas aos profissionais de saúde e ao usuário, através da bula e/ou rotulagem do produto. 3. Aprovar a seguinte norma sobre TRO. I. A composição dos produtos para a reidratação oral deve ter as seguintes especificações, de acordo com a norma internacional OMS/UNICEF: Composição por litro de água Sódio 90 meq Potássio meq Cloreto 80 meq Citrato (*) meq Glicose 111 mmol (*) O citrato pode ser substituído por bicarbonato, lactato ou acetato. Outros eletrólitos e outros componentes não aportam benefícios adicionais nesta fase de reposição e devem ser evitados, a não que surjam comprovações científicas que justifiquem sua inclusão. Não se pode adicionar corantes e/ou sabores aos produtos para reidratação oral. 183
184 Os produtos para reidratação podem ser apresentados como soluções prontas para uso, soluções concentradas para serem diluídas ou como pó ou grânulos para diluição em água, de acordo com instruções de preparo que devem constar da embalagem de forma clara como está descrito no item III desta Norma, que trata de bula e rotulagem. II. A composição dos produtos para a prevenção da desidratação e para a manutenção da hidratação deve ter as seguintes especificações : - A concentração de Sódio poderá variar entre 40 e 60 meq/l. A concentração de potássio deverá ser 20 meq/l. A concentração milimolar de bicarbonato (ou base equivalente) deverá compor 1/5 a 1/3 dos ânions, sendo o restante cloreto. A concentração de glicose deverá estar entre 110 e 140 mmol/l. - Outros eletrólitos, além dos descritos acima, poderão ser incluídos nas soluções de manutenção e prevenção de que aja justificativa de sua necessidade e das concentrações propostas. - Os produtos para prevenção da desidratação e manutenção da hidratação podem ser apresentados como soluções prontas para uso, soluções concentradas para serem diluídas ou como pó ou grânulos para diluição em água, de acordo com as instruções de preparo que devem constar da embalagem, de forma clara como está descrito no item III desta Norma, que trata de bula e rotulagem. III. A bula e a rotulagem dos produtos para hidratação oral deverão seguir as seguintes determinações. Os produtos para reidratação e para manutenção/prevenção deverão ter bula específica para cada caso, redigida de acordo com o roteiro instituído pela Portaria nº 65, de 28 de dezembro de 1984, da SNVS-MS. Mesmo os produtos em pó ou grânulos para reconstituição, vendidos em embalagem múltipla, deverão ter incluído uma bula, para orientação da farmácia e do consumidor através dos funcionários da farmácia. - O item da bula INFORMAÇÕES AO PACIENTE deve ter alto conteúdo informativo e educativo, incluindo preceitos de higiene, estímulo ao aleitamento materno, prevenção da desidratação e recuperação nutricional. É recomendável a inclusão de ilustrações. - No item da bula INDICAÇÕES, poderão ser indicados para reidratação e manutenção oral exclusivamente os produtos com teor de sódio igual a 90 meq/l. Todos os produtos com teor de sódio entre 40 e 60 meq/l somente poderão ser indicados para prevenção da desidratação e manutenção da hidratação após a fase de reidratação. - As bulas devem incluir no item PRECAUÇÕES, cuidados sobre o uso de soluções orais com potássio se a função renal estiver diminuída e, no item CONTRA INDICAÇÕES, obstrução ou perfuração intestinal. - A rotulagem dos produtos para reposição ou para manutenção, apresentados em pó, soluções concentradas para serem diluídas, ou grânulos para diluição em água, deverá conter: instruções claras de preparo, se possível com ilustrações, instruções de uso e conservação. Se dispensados em embalagem individual (envelope, sachet, etc.) deverá constar nesta, a composição do produto, bem como outros textos previstos na legislação. IV. Todos os produtos hidroeletrolíticos, cuja composição não estiver de acordo com os itens I e II desta Norma, deverão conter no rótulo em caracteres visíveis, o texto Este produto não é indicado para hidratação oral de pacientes desidratados por doença diarreica aguda. V. Os fabricantes de produtos hidratantes orais terão um prazo de 12 meses para adaptarem seus produtos ao cumprimento desta Norma. 4. Esta portaria entrará em vigor na data de sua publicação no Diário Oficial da União. 184
185 Orientações sobre medicamentos a base de água do mar 23 de abril de 2012 Os produtos a base de água do mar são matrizes complexas compostas de diversos oligoelementos cuja principal função é a limpeza da fossa nasal por meio do descongestionamento tópico. Essa atividade ocorre principalmente devido à presença de cloreto de sódio em soluções isotônicas ou hipertônicas. Tendo em vista essa característica, este tipo de produto está sujeito às normas de vigilância sanitária conforme Lei nº 6360/1976. Desta forma, devem ser registrados como medicamentos junto à Coordenação de Medicamentos Fitoterápicos e Dinamizados (COFID) na Gerência Geral de Medicamentos, conforme resolução RDC nº 24/2011. As empresas devem cumprir o Artigo 12 da Lei nº 6360/1976 que refere Nenhum dos produtos de que trata esta Lei, inclusive os importados, poderá ser industrializado, exposto à venda ou entregue ao consumo antes de registrado no Ministério da Saúde. Orientações sobre medicamentos a base de água do mar 8 de outubro de 2012 A Anvisa informa que concederá um prazo adicional de doze meses para que as empresas regularizem seus produtos a base de água do mar junto à Agência. O prazo termina em 30 de setembro de 2013, data a partir da qual nenhum produto a base de água do mar poderá ser produzido, importado, distribuído ou comercializado no país sem possuir registro na Anvisa como medicamento específico. A exigência do registro para esses produtos foi estabelecida pela Resolução RDC 24/2011. A ampliação do prazo para regularização, que vinha sendo exigida desde abril de 2012, foi uma solução que a Agência encontrou para que as empresas consigam apresentar os estudos necessários para comprovar a qualidade, segurança e eficácia dos produtos que se encontram no mercado, dando-lhes um prazo suficiente para a realização de todos os estudos necessários, como os que comprovam a estabilidade do produto. O tempo adicional também é necessário para que as empresas possam se adequar como produtoras certificadas em boas práticas de fabricação de medicamentos. A principal função dos produtos a base de água do mar é a limpeza da fossa nasal por meio do descongestionamento tópico. Essa atividade ocorre principalmente devido à presença de cloreto de sódio em soluções isotônicas ou hipertônicas. A Lei 6360/1976 estabelece que esses produtos estão sujeitos às normas de vigilância sanitária. As pessoas que utilizarem esses produtos devem ter cuidado na sua administração até a regularização dos registros. Qualquer problema de qualidade deve ser notificado à Anvisa por meio do Sistema de Notificação Notivisa. 185
186 Nota técnica: Comprovação de segurança e eficácia de vitaminas, minerais e aminoácidos 23 de abril de 2012 A RDC 24/2011, em seu Art. 33, item III, estabelece que são isentos de segurança e eficácia aqueles: medicamentos à base de vitaminas e/ou minerais e/ou aminoácidos, isolados ou associados entre si, de uso oral, classificados como medicamentos isentos de prescrição médica. A isenção de prescrição médica de um medicamento específico é determinada por dois regimentos isolados, a Portaria 40/1998 e a RDC 138/2003. Para um medicamento ser considerado isento de prescrição, a primeira premissa é que o mesmo deve ter posologia diária máxima abaixo do estabelecido pela Portaria 40/1998. Porém, nos casos em que não há limites estabelecidos nessa Portaria, a RDC 24/2011, em seu Art. 6º, parágrafo único, descreve que podem ser aplicados níveis máximos de segurança estabelecidos em outros países desde que tenham sido regulamentados através de códigos oficiais. Com relação à segunda premissa, o medicamento deve ter indicação prevista pela RDC 138/2003, devendo ser enquadrado na categoria correta e seguir conforme Art. 1º desta legislação: Todos os medicamentos cujos grupos terapêuticos e indicações terapêuticas estão descritos no Anexo: Lista de Grupos e Indicações Terapêuticas Especificadas (GITE), respeitadas as restrições textuais e de outras normas legais e regulamentares pertinentes, são de venda sem prescrição médica, a exceção daqueles administrados por via parenteral que são de venda sob prescrição médica. O medicamento só pode ser considerado isento de prescrição se estiver categorizado corretamente. Tendo em vista o disposto e de modo ilustrativo, seria errôneo isentar de comprovação de eficácia um medicamento cujo ativo seja colecalciferol e a categoria e indicação propostas sejam antidiarréico e diarréia, respectivamente. 186
187 Adequação das Soluções Parenterais de Grande Volume SPGV ao sistema fechado. As Instituições de Saúde de acordo com a RDC 45/03 deverão substituir o sistema de infusão aberto para o fechado até a data de Março Para que ocorra está substituição as SPGV deverão alterar o sistema de fechamento do frasco/bolsa para o sistema fechado. O Sistema fechado é definido como a administração de Solução Parenteral na qual durante todo o preparo e administração, não permite o contato da solução com o meio ambiente. As empresas deverão adequar os frascos/bolsas dos medicamentos seguindo a RDC 29/07. A referida resolução estabelece que os medicamentos ostentem em seus rótulos a seguinte frase: SISTEMA FECHADO. Abaixo tem a ilustração da alteração a ser efetuada na embalagem: 187
188 Seminário de Orientação ao Setor Regulado na Área de Medicamentos Respostas aos questionamentos referentes às apresentações das categorias de Medicamentos Específicos e Fitoterápicos 1. Como cumprir a exigência de apresentar a foto da CCD (cromatografia em camada delgada) no tempo inicial da estabilidade e no tempo final, depois que o estudo já está concluído? Estes dados deveriam estar disponíveis na empresa para a inspeção. R. Podem ser apresentados não só fotografias, mas qualquer forma de registro da imagem inicial do CCD; formas confiáveis evidentemente excluem desenhos representativos dos cromatogramas. Mesmo que o estudo já esteja terminado, se foram usados CCD s estes evidentemente devem ter sido registrados. 2. A portaria 185/99 afirma que o importador deve relatar os ensaios a serem realizados no país, mas não afirma quais. Consultamos a inspeção, que demonstrou não haver consenso sobre o que exigir. A princípio todos pensam que o doseamento é fundamental a identificação também. Se a ANVISA deseja que todos os ensaios farmacopéicos sejam refeitos quando o produto entra no Brasil, deve regulamentar esta exigência tornando claro as obrigações do importador? R. É necessário a realização completa de todos os testes de controle de qualidade do produto conforme Portaria 185/99: Art. 2º A empresa importadora é responsável pela qualidade, eficácia e segurança dos produtos que importar. 1º Todos os produtos importados devem ser submetidos a ensaios completos de controle de qualidade, lote a lote, através de laboratório próprio da importadora ou da contratação de serviços de terceiros. 3. Esclarecer se é necessário realizar analise de controle de qualidade completa para produto acabado importado ou se a realização do teste de doseamento e outros testes mais importantes atendem as disposições da GMEFH. R. É necessário a realização completa de todos os testes de controle de qualidade do produto conforme Portaria 185/99: Art. 2º A empresa importadora é responsável pela qualidade, eficácia e segurança dos produtos que importar. 1º Todos os produtos importados devem ser submetidos a ensaios completos de controle de qualidade, lote a lote, através de laboratório próprio da importadora ou da contratação de serviços de terceiros. 4. A metodologia analítica de controle de qualidade foi desenvolvida e validada em outro país. Para realizar a analise de rotina tem que fazer a co-validação. Por ser importado no Brasil não tem como conseguir o placebo e o ativo? O que fazer? R. Nos casos de transferência de metodologia analítica de controle de qualidade a empresa deverá realizar a covalidação de acordo com a RE 899/03 que diz: 1.8. No caso da transferência de metodologias da matriz para suas subsidiárias no Brasil e/ou das empresas nacionais para os centros de estudos de equivalência farmacêutica, a metodologia será considerada validada, desde que sejam avaliados os parâmetros de precisão, especificidade e linearidade. Cópia de toda a documentação original da validação da metodologia deverá ser anexada, como prova de que a metodologia foi originalmente validada e deverá conter, no mínimo, todos os parâmetros relacionados no item 1.7. O fato de ser importado não justifica a afirmativa de que não se pode conseguir o ativo e o placebo. Se realmente não for possível consegui-los, a falta da co-validação impedirá a concessão do registro. 5. Para adequação à categoria de específicos, mesmo que o produto não tenha alterado a formulação, é necessário a realização de novo estudo de estabilidade? Por exemplo, uma vitamina C que antes era registrada como similar, no momento da renovação passa a ser classificada como específico. Há necessidade de apresentar novos estudos? R. Não desde que obedecido os critérios de adequação presentes da RE 01/
189 6. Na apresentação de específicos, ficou claro que nos casos de polivitamínicos devem-se analisar todas as vitaminas que compõem o produto. No entanto foi falado anteriormente que em alguns casos (exemplo ácido fólico) escolhe-se apenas um ativo para realizar o doseamento. Gostaria de esclarecimento sobre qual o procedimento correto a ser adotado. R. A empresa deve realizar o controle de qualidade (doseamento) de todas as substâncias ativas de sua formulação, e no caso do teste de dissolução de comprimidos polivitamínicos e poliminerais a empresa deverá realizar o doseamento de apenas um ativo. 7. Como estão sendo conduzidas as adequações de marcas que já estão no mercado, com relação ao complemento PLUS, MAX, HIPER? Na renovação as empresas deverão retirar esses complementos de marca? R. Sim, pois de acordo com o artigo 5, 1, 3 da lei nº 6360/1976 e Resolução RDC n 92/2000 em seu artigo 4, 2 e 7, esta Agência entende que os radicais PLUS, MAX e HIPER não poderão ser adotados uma vez que induzem o consumidor a acreditar que o medicamento tenha um efeito terapêutico maior que outro medicamento com mesmos princípios ativos e concentrações, o que não é característica do produto em questão. E ainda, a Portaria 34 de 20/12/66 em seu item primeiro diz: Não será permitido o uso das expressões concentrado, forte, fraco e de outras com o mesmo propósito para identificação de especialidade farmacêutica apresentada em diferentes concentrações. 8. Vitaminas em produtos isentos de prescrição em que o paciente tem conhecimento dos chamados nomes populares seria possível adotarmos, além da DCB, seu nome popular? R. Sim, pois de acordo com o artigo 3.5 do anexo da RDC 333/03 diz: No caso de polivitamínicos, poliminerais e ou poliaminoácidos pode-se adotar os sinônimos usuais apresentados na literatura técnica. Os nomes ainda podem estar acrescidos de uma sigla, complemento adicional ou parte do nome do laboratório de reconhecida propriedade do mesmo. (grifo nosso) 9. No processo a empresa apresenta estudos de estabilidade realizados de acordo com RE 560/02 (com lotes de fabricação datados de 2003). A empresa pode apresentar estes estudos no registro e/ou renovação? R. Sim, pois de acordo com a RE 01/05 no seu item 5.2 diz: A Anvisa aceitará até 31 de julho de 2007, no momento do registro, pós-registro ou da renovação de registro, estudos de estabilidade de longa duração que já estejam em andamento com o parâmetro de umidade abaixo de 75%. Entretanto as câmaras climáticas devem ser re-qualificadas para umidade de 75% a partir da data de publicação desta Resolução. Fica a critério da empresa reiniciar ou não estes estudos. Salientamos que se aplica aos estudos realizados com o parâmetro de umidade diferente, como no caso da RE 560/ Participei no ano passado, de várias discussões com relação a RDC 138/03 e nestas discussões com a própria ANVISA, era previsto que nos casos de associação poderia se enquadrar o medicamento em mais de um grupo terapêutico e com isso usar as indicações desses grupos. Exemplo: produto BBB (principio ativo A + princípio ativo B). Os dois ativos estão enquadrados no GITE e, portanto, poderiam ser utilizadas as indicações do grupo terapêutico onde está enquadrado o principio ativo A + indicação do grupo terapêutico onde está enquadrado o principio ativo B. Pergunta: Este entendimento está correto? Esse entendimento foi alterado dentro da Anvisa? R. O produto poderá ou não ser enquadrado em duas categorias presentes na GITE, dependendo das propriedades terapêuticas alegadas para o medicamento, considerando-se a racionalidade da associação comprovada através de estudos clínicos que serão analisados caso a caso. 189
190 11. É impraticável, muitas vezes, utilizar padrão na CCD já que a quantidade necessária é muito grande e os padrões normalmente são caros, e só comercializados em pequenas quantidades. Não podemos realizar a CCD de uma referência bibliográfica reconhecida? Se não, por quê? R. As quantidades requeridas para a execução de CCD são mínimas, portanto o argumento de alto custo não se sustenta. Pode ser admitida a apresentação de CCD do extrato, comparado a uma CCD padrão de uma referência bibliográfica reconhecida; a aceitação dependerá da qualidade tanto da referência como da CCD do extrato. 12. A exemplo da Valeriana officinalis, cuja monografia consta em farmacopéia e nesta exige a análise do marcador ácido valerênico, considerando que é o marcador que garante a qualidade do produto, pergunta: Por que, não aceitar a realização de análise de acordo com essa farmacopéia? R. A questão não tem muito sentido, uma vez que a questão não é aceitar ou deixar de aceitar o que está na Farmacopéia: o que acontece é que a Farmacopéia Européia e a USP diferem na forma de dosar o extrato, a européia dosa todos os ácidos valerênicos, a USP apenas o ácido valerênico. Ambos são aceitos, desde que, no caso de doseamento feito de acordo com a USP, se demonstre a correspondência com o teor total de ácidos valerênicos, uma vez que é esse padrão (teor de ácidos valerênicos totais) que está presente na literatura sobre a eficácia e a segurança do produto. 190
191 1. Introdução Guia para a condução de estudos não clínicos de toxicologia e segurança farmacológica necessários ao desenvolvimento de medicamentos O presente Guia é uma orientação para a condução de estudos não clínicos de segurança durante o desenvolvimento de medicamentos. Porém, caso o pesquisador/instituição consiga comprovar a segurança desses fármacos por outros estudos científica e tecnicamente mais viáveis, os dados apresentados poderão ser avaliados pela ANVISA. O uso de métodos alternativos in vitro em substituição a estudos in vivo, desde que validados e aceitos internacionalmente são recomendados. A sua elaboração foi baseada em documentos de agências reconhecidas pela vigilância sanitária de mmedicamentos (FDA, EMA), e de instituições de interesse na área (ICH, OECD, NCI, WHO), visando uma maior harmonização com a regulamentação internacional. Além disso, o guia também tem a intenção de racionalizar estudos não clínicos, evitando duplicidades e utilização desnecessária de animais sem que isso possa comprometer a obtenção e a confiabilidade de informações referentes à segurança da substância a ser testada. Os estudos não clínicos de segurança propostos nesse documento incluem: estudos de toxicidade de dose única (Aguda), toxicidade de doses repetidas, toxicidade reprodutiva, genotoxicidade, tolerância local e carcinogenicidade além de estudos de interesse na avaliação da segurança farmacológica e toxicocinética (Administração, Distribuição, Metabolismo e Excreção ADME). Outros estudos que avaliam a segurança da substância teste poderão ser necessários conforme o caso. Os estudos propostos devem ser conduzidos de acordo com as Boas Práticas de Laboratório (BPL) - OECD Principles of Good Laboratory Practice; HANDBOOK: GOOD LABORATORY PRACTICE (GLP) /WHO (Quality practices for regulated non-clinical research and development), quando aplicável, e os animais a serem utilizados deverão ser saudáveis, preferencialmente livres de patógenos (SPF Specific Pathogen Free), e de origem conhecida, além de possuir peso e idade adequados ao experimento. Em caso de fármacos com novo mecanismo de ação, recomendamos que seja previamente avaliado se a espécie animal indicada para a realização dos estudos de segurança é considerada a mais apropriada para a extrapolação de dados para humanos. Para novos excipientes devem ser conduzidos estudos não clínicos de segurança, caso não haja informações sobre sua toxicidade. Ressaltamos que o desvio das orientações do guia deverá sempre ser justificado. A partir do guia pretende-se que os estudos não clínicos de segurança necessários o desenvolvimento de novos medicamentos sejam realizados de forma harmonizada e cientificamente válida. Além disso, espera-se que eles possam fornecer dados confiáveis para dar subsídios às futuras Pesquisas Clínicas. 2. Estudos de Toxicidade de Dose Única (Aguda) Definição/Objetivo: Os estudos de toxicidade aguda são aqueles utilizados para avaliar a toxicidade produzida por uma substância teste quando esta é administrada em uma ou mais doses durante um período não superior a 24 horas, seguido de observação dos animais por 14 dias após a administração. Modelo animal a ser estudado: Devem ser conduzidos com no mínimo duas espécies de mamíferos. Via de administração: Utilizar duas vias de administração: (1) a pretendida para administração em humanos e (2) a parenteral. Se a administração endovenosa for a pretendida para uso em humanos, a utilização de apenas esta via para estudos de toxicidade de dose única é suficiente. 191
192 Quando a via pretendida para uso em humanos for a oral, é recomendável a administração do produto em animais por gavagem. Dosagem: A dose limite a ser testada será de 1000 mg/kg/dia para roedores e não roedores. Em situações, em que essa dose não resulte em uma margem de 10 vezes a exposição clínica e a dose clínica exceda 1 grama por dia, deve ser considerada a menor dose disponível entre 10 vezes a exposição clínica, 2000 mg/kg/dia ou a máxima dose disponível. Período de Observação: Período de observação dos animais: no mínimo 14 dias após a administração da substância teste. No dia da administração os animais devem ser observados no mínimo duas vezes. Posteriormente, no mínimo uma vez ao dia. Observações: Estes estudos devem ser realizados anteriormente à Fase I da Pesquisa Clínica. Estudos para a determinação de DL50 (dose letal 50% - dose que mata 50% dos animais) não são necessários. Podem ser utilizados métodos alternativos para a estimativa da dose letal envolvendo um menor número de animais, tais como os preconizados nos guias da OECD. 3. Estudos de Toxicidade de Doses Repetidas Definição/Objetivo: Os estudos de toxicidade de doses repetidas têm como objetivo, caracterizar o perfil toxicológico da substância pela administração repetida. A partir deles é possível a obtenção de informações sobre os efeitos tóxicos, identificação de órgãos alvos, efeitos na fisiologia do animal, hematológicas, bioquímicas, anátomo e histopatológicas, além de informações sobre a indicação do NOEL e NOAEL. Modelo animal a ser estudado: Devem ser conduzidos com no mínimo duas espécies de mamíferos, incluindo uma espécie não roedora. A amostra deve contemplar números iguais de machos e fêmeas (a utilização de apenas um dos sexos deve ser justificada pela indicação). As espécies devem ser selecionadas com base em sua relevância para a extrapolação de dados para seres humanos, considerando a farmacocinética, farmacodinâmica e biodisponibilidade da substância teste, incluindo sua biotransformação. Via de administração: Deverá ser utilizada a via em que a droga será administrada em humanos, mas se a absorção em animais for limitada em relação ao homem, também uma via parenteral. Dosagem: As doses utilizadas em estudos de administrações repetidas geralmente são estabelecidas a partir das informações produzidas em estudos de toxicidade aguda ou testes piloto para indicação de doses. Geralmente 3 doses são utilizadas, sendo a mais alta escolhida com a expectativa de produzir efeitos tóxicos observáveis, mas não morte nem sofrimento intenso e respeitando-se o limite máximo de 1000 mg/kg/dia em roedores e não-roedores ou as situações particulares discutidas no item dosagem dos estudos de toxicidade de dose única. As demais doses são estabelecidas em seqüencia descendente sugerindo-se intervalos de 2 a 4 vezes. Período de Observação Duração Mínima dos estudos de Toxicidade de Doses Repetidas: Vide quadro abaixo*. 192
193 *ICH Harmonised Tripartite Guideline: Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals M3(R2)- junho/2009. Parâmetros a serem avaliados: Roedores: Mortalidade, sinais clínicos (incluindo parâmetros comportamentais); variações no peso corporal e no consumo de ração e água, patologia clínica (hematologia, bioquímica); duração e reversibilidade da toxicidade; investigações anátomo e histopatológicas. Não Roedores: Mortalidade; sinais clínicos (incluindo parâmetros comportamentais); variações no peso corporal e no consumo de ração e água; patologia clínica (hematologia, bioquímica); oftalmologia; duração e reversibilidade da toxicidade; investigações anátomo e histopatológicas. Observações: Os dados de segurança obtidos nesses estudos dão suporte às Fases 1, 2 e 3 da Pesquisa Clínica. As condições ambientais devem ser as preconizadas para a espécie estudada. Deve-se monitorar a qualidade e composição da dieta e água durante todo o período do estudo e recomenda-se registrá-las no relatório. Para avaliação do potencial de toxicidade de doses repetidas podem ser utilizados os guias da OECD. 4. Estudos de Toxicidade Reprodutiva Definição/Objetivo: O objetivo dos estudos de toxicidade reprodutiva é revelar algum efeito de uma ou mais substâncias ativas na reprodução de mamíferos. Para este propósito, investigações e interpretações dos resultados devem ser relacionadas com outros dados farmacológicos e toxicológicos disponíveis, para determinar situações em que riscos potenciais para a reprodução humana são maiores, menores ou iguais àqueles relativos a outras manifestações toxicológicas. Estudos a serem realizados: Os estudos de toxicidade reprodutiva devem contemplar principalmente avaliações nas seguintes fases: A. Fertilidade e desenvolvimento embrionário inicial, B. Desenvolvimento pré e pós-natal, incluindo função materna, C. Desenvolvimento embrio-fetal. Existe a possibilidade de agrupar as avaliações com roedores em um ou dois estudos. Na abordagem de estudo único, a administração deve se iniciar antes do acasalamento e persistir até o nascimento. 193
194 A abordagem de dois estudos mais simples consiste em realizar o estudo de fertilidade e associar o estudo de desenvolvimento pré e pós natal com o estudo de desenvolvimento embrio-fetal. Porém, devem-se realizar os estudos de embriotoxicidade também em uma espécie não roedora. Para avaliação do potencial de toxicidade reprodutiva podem ser utilizados os guias da OECD. A -Fertilidade e desenvolvimento embrionário inicial A.1. Modelo animal a ser estudado: No mínimo uma espécie roedora, preferencialmente ratos. A proporção adequada de machos e fêmeas é de 1:1. A.2. Via de Administração: A mesma pretendida para uso humano. A.3. Dosagem: A seleção de doses é um dos pontos mais críticos nos desenhos dos estudos de toxicidade reprodutiva. A escolha da dose alta deve ser baseada nos dados de todos os estudos disponíveis (farmacologia, estudos de toxicidade aguda/ crônica e toxicocinética). Uma vez determinado a dose alta, as demais doses devem ser selecionadas na seqüência descendente e os intervalos entre elas dependem da cinética e de outros fatores de toxicidade. O período de administração poderá ser embasado por dados de estudos de toxicidade de doses repetidas de, no mínimo, um mês de duração. Caso não sejam observados efeitos tóxicos no sistema reprodutor, um intervalo de tratamento pré-acasalamento de 2 semanas para fêmeas e 4 semanas para machos pode ser utilizado. O tratamento deve ocorrer desde o acasalamento e continuar até a eutanásia para os machos e no mínimo até a fase de implantação para as fêmeas. A.4. Período de Observação: Fêmea: Período fértil, implantação e desenvolvimento dos estágios embrionários de préimplantação. Macho: Fase adulta para avaliação de efeitos funcionais (por exemplo, libido, maturação do esperma epididimal) que possam não ter sido detectados em exames histológicos do órgão reprodutor. A.5. Parâmetros a serem avaliados: Avaliações: maturação de gametas, comportamento no acasalamento, fertilidade, estágio de pré-implantação embrionária, implantação. Durante o estudo: sinais clínicos e mortalidade, alteração de peso corpóreo, consumo de ração, esfregaços vaginais diariamente, pelo menos durante o período de acasalamento, observações relevantes provenientes de outros estudos de toxicidade. Ao Final do Estudo: Necropsia de todos os adultos, preservar órgãos com alterações macroscópicas e órgãos controle para posterior avaliação; preservar testículos, epidídimo, ovários e útero de todos os animais para possível avaliação histológica; contagem e viabilidade de esperma em epidídimo ou testículo; contagem de corpos lúteos e implantações; fetos vivos e mortos. B -Desenvolvimento pré e pós-natal, incluindo função materna: B.1. Modelo animal a ser estudado: No mínimo uma espécie roedora, preferencialmente ratos. B.2. Via de Administração: A mesma pretendida para uso humano. Fêmeas devem ser expostas à substância teste desde a implantação até o final da lactação. B.3. Dosagem: A seleção de doses é um dos pontos mais críticos nos desenhos dos estudos de toxicidade reprodutiva. A escolha da dose alta deve ser baseada nos dados de todos os estudos disponíveis (farmacologia, estudos de toxicidade aguda/crônica e toxicocinética). Uma vez determinada a dose alta, as demais doses devem ser selecionadas na sequência descendente e os intervalos entre elas dependem da cinética e de outros fatores de toxicidade. 194
195 B.4. Período de Observação: Período de prenhez e lactação das fêmeas avaliadas. B.5. Parâmetros a serem avaliados: Avaliações: Aumento da toxicidade relativa a fêmeas não prenhes, mortalidade pré e pós-natal dos filhotes, crescimento e desenvolvimento alterados, alterações funcionais dos filhotes, incluindo comportamento, maturidade (puberdade) e reprodução. Durante o estudo: sinais clínicos e mortalidade, alteração de peso corpóreo, observações relevantes provenientes de outros estudos de toxicidade, duração da prenhez e parição. Ao Final do Estudo: Necropsia e avaliação macroscópica de todos os adultos, implantações, anormalidades. Contagem de fetos vivos e mortos e peso corpóreo ao nascimento, sobrevivência / crescimento / peso corporal pré e pós-lactação, maturação e fertilidade, desenvolvimento físico, funções sensoriais e reflexas, comportamento da ninhada. B.6. Observações: Um macho e uma fêmea por ninhada devem ser selecionados para acasalamento na idade adulta com objetivo de avaliar a sua competência reprodutiva. C -Desenvolvimento embrio-fetal: C.1. Modelo animal a ser estudado: Usualmente duas espécies: uma roedora, preferencialmente ratos, e uma não roedora, preferencialmente coelhos. O uso de outras espécies deverá ser justificado. C.2. Via de Administração: A mesma pretendida para uso humano. C.3. Dosagem: A seleção de doses é um dos pontos mais críticos nos desenhos dos estudos de toxicidade reprodutiva. A escolha da dose alta deve ser baseada nos dados de todos os estudos disponíveis (farmacologia, estudos de toxicidade aguda/ crônica e toxicocinética). Uma vez determinada a dose alta, as demais doses devem ser selecionadas na sequência descendente e os intervalos entre elas dependem da cinética e de outros fatores de toxicidade. C.4. Período de Observação: Fêmeas devem ser submetidas à eutanásia e examinadas um dia antes da parição. Todos os fetos devem ser identificados individualmente e examinados quanto à viabilidade e anormalidades no dia da eutanásia. C.5. Parâmetros a serem avaliados: Durante o estudo: sinais clínicos e mortalidade, alteração de peso corpóreo, consumo de ração,observações relevantes provenientes de outros estudos de toxicidade. Ao final do estudo: necropsia com avaliação macroscópica de todos os adultos, contagem de corpos lúteos, número de implantações que resultaram em fetos vivos e mortos, peso corpóreo individual fetal, anormalidades fetais, avaliação da placenta, preservar órgãos com achados macroscópicos para possíveis avaliações histopatológicas e órgãos correspondentes em quantidade suficiente para comparação (controle). C.6. Observações: Nos estudos é necessário que os animais tenham idade e peso comparáveis. Lactação: Em casos de avaliação de riscos para lactentes (cujas mães foram expostas à substância teste durante a lactação), deverão ser considerados dados não clínicos, farmacocinéticos, e às vezes clínicos. As avaliações não clínicas são compostas de: transferência para o leite, desenvolvimento dos filhotes lactentes, características fisicoquímicas e farmacocinéticas da substância, quantidade absorvida estimada e permanência da substância no leite. 195
196 Considerações sobre os estudos de toxicidade reprodutiva que devem ser concluídos anteriormente à inclusão de homens, mulheres sem e com potencial para engravidar nas fases de Pesquisa Clínica: Homens: podem ser incluídos em Pesquisas Clínicas Fase 1 e 2 anteriormente à condução de estudos de fertilidade em machos desde que uma avaliação dos órgãos de reprodução já tenha sido realizada nos estudos de toxicidade de doses repetidas. Estudos de fertilidade em animais machos devem ser concluídos anteriormente à iniciação de Pesquisas Clínicas Fase 3. Mulheres sem potencial para engravidar (em menopausa há pelo menos 1 ano, estéreis permanentes): podem ser incluídas em Pesquisas Clínicas sem estudos de toxicidade reprodutiva desde que tenha ocorrido a avaliação dos órgãos reprodutivos nos estudos de toxicidade de doses repetidas. Mulheres com potencial para engravidar, utilizando métodos contraceptivos podem ser incluídas em Estudos Clínicos de fase I e II sem os estudos de toxicidade de desenvolvimento (por exemplo, embriotoxicidade) desde que se configure uma das circunstâncias citadas abaixo: 1- Administração da substância teste em estudo por até duas semanas sob intensivo controle do risco de gravidez (por meio de: teste de gravidez βhcg, métodos de controle altamente eficazes e inicialização do estudo após período menstrual confirmado), ou 2- Predominância da doença em mulheres e o objetivo do estudo clínico não pode ser efetivamente conhecido sem a inclusão dessa população e há intensivo controle do risco de gravidez conforme métodos apresentados acima, ou 3- Conhecimento do mecanismo de ação da substância teste, tipo de substância teste, extensão da exposição fetal ou dificuldade da condução de estudos de toxicidade de desenvolvimento em modelo animal apropriado, ou 4- Existência de dados preliminares de toxicidade reprodutiva em duas espécies, com intensivo controle do risco de gravidez conforme métodos apresentados acima, em estudos de curta duração (até 3 meses) e até 150 sujeitos de pesquisa. Para as demais situações, os estudos de embriotoxicidade devem estar concluídos antes da inclusão dessa população em estudos clínicos. As mulheres com potencial para engravidar podem ser incluídas nos estudos clínicos iniciais (Fase 1 e 2) anteriormente à condução de estudos de fertilidade desde que tenham sido realizados avaliação de órgãos reprodutivos em estudos de toxicidade de doses repetidas. Os estudos de desenvolvimento pré e pós-natal deverão ser finalizados e apresentados à autoridade reguladora no dossiê do processo de registro do medicamento. 5. Estudos de Genotoxicidade Definição/Objetivo: Os estudos de genotoxicidade são testes in vitro e in vivo desenhados para detectar o potencial das substâncias sob investigação de causar mutações gênicas e alterações cromossômicas. Estudos a serem realizados: Opção 1: Bateria de testes: 1- Um teste para mutação gênica em bactéria, 2- Um teste citogenético para avaliação de dano cromossômico (teste de aberrações cromossômicas in vitro ou teste de micronúcleo in vitro) ou um teste in vitro de mutação gênica em células tk de linfoma de camundongo, 3- Um teste in vivo para genotoxicidade, geralmente um teste de dano cromossômico em células hematopoiéticas de roedores, podendo ser micronúcleos ou aberrações cromossômicas. Opção 2: Bateria de testes: 1- Um teste para mutação gênica em bactéria, 2- Uma avaliação de genotoxicidade in vivo em dois tecidos, geralmente um teste de micronúcleo em células hematopoiéticas de roedores e um segundo ensaio in vivo. Modelos Biológicos a serem Estudados: 196
197 Para o teste de mutação gênica recomenda-se a utilização de linhagens de Salmonella typhimurium para detectar mudanças nos sítios de Guanina-Citosina (G-C) como TA1535, TA1537 (ou TA 97, ou TA97a), TA98 e TA100. Para a detecção de pontos de mutação nos sítios de Adenina-Timina (A-T) deve-se utilizar Salmonella typhimurium TA 102, Escherichia coli WP2 uvra ou Escherichia coli WP2 uvra (pkm101). Os testes de aberrações cromossômicas em células hematopoiéticas de roedores podem detectar um amplo espectro de mudanças na integridade cromossômica. Os ensaios in vitro devem ser realizados sempre em duas condições: na presença e na ausência de ativação metabólica. Para o teste de micronúcleos in vivo recomenda-se a utilização de roedores (camundongos ou ratos), apenas um sexo, de preferência machos, desde que não haja dados que indiquem a discrepância de toxicidade entre os sexos. Quando substâncias sexo específicas estão sendo testadas, o ensaio deverá ser realizado no sexo apropriado. Via de Administração (quando aplicável): Similares àquelas preconizadas para uso humano. Dosagem (quando aplicável): Para teste de Ames: Para bactérias, as concentrações máximas recomendáveis são de 5 mg/placa, quando não limitado por solubilidade ou citotoxicidade. Compostos com difícil solubilização têm sua concentração máxima de exposição limitada a sua solubilidade em veiculo compatível com o sistema testado. Para células de mamíferos: As concentrações máximas recomendáveis são de 1,0 mm ou 0,5 mg/ml, o que for menor, quando não limitado por solubilidade ou citotoxicidade. Para testes in vivo: Protocolos de curta duração: (geralmente 1-2 administrações): a máxima dose recomendada é uma dose limite de 2000 mg / kg se esta é tolerada, ou a máxima dose em que níveis mais altos seria esperado para a produção de letalidade. Doses mais baixas são geralmente espaçadas em aproximadamente dois a três intervalos de vezes inferiores a essa dose. Protocolos de longa duração (Estudos de Administração Múltipla): Três níveis de doses sendo que a dose máxima deve ser: a máxima dose tolerada, 1000 mg/kg para estudos de 14 dias ou maiores, se esta dose é tolerada ou a dose que leva à saturação da exposição. Parâmetros a serem avaliados: Os testes acima devem ser capazes de revelar resultados claramente positivos ou negativos, entretanto, alguns dos resultados dos testes acima não podem ser apresentados na forma de positivo ou negativo sob critérios prédeterminados, portanto são declarados inconclusivos após a aplicação de interpretações estatísticas e interpretação biológica adequada. Em casos inconclusivos ou fracamente positivos pode haver a necessidade da repetição do ensaio, eventualmente com a modificação do protocolo como, por exemplo, o espaçamento dos níveis das concentrações. Os testes de genotoxicidade devem ser capazes de avaliar os potenciais danos ao DNA que podem ser observados na forma de mutações gênicas e alterações cromossômicas, que podem ser numéricas ou estruturais. Estas podem estar fortemente relacionadas à produção de alterações hereditárias e em diferentes etapas do desenvolvimento de malignidades. Compostos que apresentam resultados positivos nos testes supracitados são potencialmente agentes carcinogênicos e/ou mutagênicos para seres humanos. Apesar da relação entre exposição a agentes químicos e carcinogênese se encontrar estabelecida em humanos, tem sido difícil a avaliação da transmissão hereditária de alterações provocadas por tais agentes, portanto os testes de genotoxicidade têm sido utilizados, principalmente, para a previsão de potencial carcinogênico. 197
198 Observações: Um ensaio de mutação gênica é geralmente considerado suficiente para dar suporte a todos os estudos clínicos de administração única. Estudos com doses múltiplas necessitarão de suporte de pelo menos um dos dois conjuntos de testes descritos como opção 1 e opção 2. Os testes de genotoxicidade devem estar concluídos anteriormente à realização das Pesquisas Clínicas fase 2. Para avaliação do potencial de genotoxicidade podem ser utilizados os guias da OECD. 6. Estudos de Tolerância Local Definição/Objetivos: A avaliação de tolerância local deve ser realizada em testes de laboratório antes da exposição humana ao produto. O objetivo destes estudos é saber se as substâncias (princípios ativos e excipientes) são toleradas em locais do corpo que poderão entrar em contato com o produto em conseqüência da sua administração na prática clínica. Os testes deverão avaliar quaisquer efeitos mecânicos da administração ou ações meramente físico-químicas do produto que podem ser distinguidas de efeitos toxicológicos ou farmacodinâmicos. É preferível avaliar a tolerância local pela via terapêutica pretendida como parte dos estudos de toxicidade geral; estudos autônomos geralmente não são recomendados. Modelo animal a ser utilizado: A escolha da espécie dependerá do problema a ser investigado e ao modelo considerado adequado. Normalmente, apenas uma espécie deve ser avaliada para cada tipo de ensaio. Onde dois ou mais parâmetros toxicológicos estão sendo investigados (por exemplo, tolerância ocular e sensibilização da pele), as espécies adequadas para cada ensaio deverão ser utilizadas. Dosagem: Regra geral, a real concentração de substância ativa utilizada em seres humanos deve ser testada. A dose pode, então, ser ajustada através da variação da freqüência de administração. Administração: Ensaios de tolerância local devem ser realizados com a preparação desenvolvida para utilização no ser humano, utilizando o veículo e/ou os excipientes no tratamento do grupo controle. Controles positivos/ substância referência pode ser incluída, sempre que necessário. A freqüência e a duração da administração aos animais deverão ser determinadas pela proposta de administração, em condições de uso clínico. Contudo, o período de aplicação não deve ser superior a quatro semanas. Investigação de tolerância local após a administração acidental pode ser feita por estudos de dose única. Via de Administração: A via de administração deve ser selecionada de acordo com o previsto para o ser humano. A anatomia e fisiologia do local de aplicação devem ser consideradas. Testes em diferentes vias de administração no mesmo animal são permitidos, desde que os testes não influenciem uns aos outros, e na medida em que permite tolerância sistêmica. Administração contralateral da preparação controle é aceitável quando a confiabilidade do estudo não for comprometida. Reversibilidade: Avaliações de reversibilidade das lesões locais deverão ser incluídas quando relevantes. Bem-estar animal: Em caso de forte irritabilidade em testes com modelos menos sensíveis, o progresso a modelos mais sensíveis deve ser cuidadosamente considerado. Cuidados também devem ser tomados para minimizar a exposição dos animais a irritações dolorosas, encerrando os experimentos no momento apropriado, ou seja, no ponto onde são vistas reações adversas graves e não esperar a continuação para fornecer resultados essenciais para a avaliação dos riscos. 198
199 Estudos Realizados: A. Testes de Tolerância no Local de Administração B. Teste de Toxicidade Sistêmica C. Testes de Tolerância para Vias Específicas de Administração D. Potencial de Sensibilidade A. Testes de Tolerância no Local de Administração Os testes de Tolerância Local devem ser realizados nos locais que entrarão em contato com a substância como resultado do método de administração e também em locais que poderão entrar em contato acidentalmente ou devido à exposição inevitável ao produto. O local de administração pode ser o mesmo órgão ou tecido, que se destina a ser o alvo terapêutico (por exemplo, a pele para produtos dermatológicos, o olho para medicamentos oftalmológicos), ou o local de administração afastados do alvo terapêutico (Por exemplo, sistemas transdérmicos, medicamentos administrados por via endovenosa). No planejamento de ensaios de tolerância local, as seguintes informações devem consideradas: As propriedades físico-químicas do produto. Por exemplo, para evitar sofrimentos desnecessários, produtos que são corrosivos pelo seu ph devem ser excluídos de estudos em animais vivos. A farmacodinâmica, dados toxicológicos e farmacocinéticos para a substância ativa, ou a combinação de substâncias ativa, bem como dos excipientes. Avaliação global dos resultados deverá incluir a discussão sobre a adequação do teste de tolerância local e sobre o significado dos resultados para a utilização clínica do produto. O teste de tolerância local pode ser parte de outros estudos sobre toxicidade desde que o medicamento seja administrado sob condições adequadas. B. Teste de Toxicidade Sistêmica Nos casos em que o medicamento não foi testado anteriormente, estudos de avaliação de toxicidade deverão ser conduzidos. Estes estudos devem empregar uma via de administração adequada, que resulta na exposição sistêmica. O ensaio deve incluir as investigações necessárias para revelar sinais de toxicidade em órgãos alvo. Geralmente, estudos para a avaliação da toxicidade sistêmica não serão exigidos nos casos em que: Absorção do produto é tão baixa que a possibilidade de efeitos sistêmicos pode ser excluída; O produto é absorvido, mas a sua toxicidade sistêmica foi previamente investigada. C. Testes de Tolerância para Vias Específicas de Administração Via dérmica: Modelo animal a ser utilizado: A seleção das espécies deve ser justificada, geralmente são utilizados coelhos. Parâmetros a serem Avaliados: Em relação aos testes de tolerância dérmica, devem ser considerados, no mínimo: testes de dose única, doses repetidas e avaliação do potencial de sensibilidade. -Para testes de dose única: 1-Testes relacionados à corrosão da pele poderão ser realizados in vitro. 2-Testes de irritação poderão ser realizados em coelhos. A pele é examinada para avaliar o grau de eritema, edema, descamação, formação de cicatriz e outras lesões. A duração do estudo dependerá das mudanças observadas em 24, 48 e 72 horas depois da administração. Caso as mudanças persistam, poderão ser necessárias observações por período maior que oito dias após a administração. -Para testes de doses repetidas: 1-Deverão ser conduzidos em coelhos (outras espécies animais podem ser utilizadas se adequadamente validadas). 2-A pele deverá ser avaliada durante o mesmo período citado para os testes de dose única. 3-Exames histológicos devem ser considerados caso a caso. 199
200 -Potencial de Sensibilidade: Sistemas de testes recomendados: Ensaio em cobaia, Ensaio de Linfonodo Local. Via Parenteral: Modelo animal a ser utilizado: A seleção das espécies deve ser justificada. Parâmetros a serem Avaliados: -Freqüência de administração: Única. Em alguns casos, administrações repetidas (sete dias) podem ser importantes. -Para única administração, observações repetidas dos animais e dos sítios de injeção devem ser realizadas durante horas depois da administração. Depois disso, um cuidadoso exame macroscópico dos sítios de injeção e tecidos circundantes deve ser realizado. Caso seja necessário para diagnóstico, um exame histológico poderá ser também realizado. -Para administrações repetidas, observações dos sítios de injeção e de animais devem ser realizadas. Depois da última administração, os procedimentos devem ser os mesmos recomendados para administração única. Via ocular: Modelo animal a ser utilizado: A seleção das espécies deve ser justificada, geralmente são utilizados coelhos. Parâmetros a serem Avaliados: O tipo de ensaio de tolerância ocular será determinado pela magnitude da exposição ao produto (por exemplo, produtos que se destinem a ser administrados repetidamente no olho, obviamente requerem estudos mais extensos do que produtos sujeitos a exposição acidental). A avaliação da tolerância para os medicamentos destinados a administração ocular exige: Um teste de tolerância ocular de dose única (normalmente realizado em coelhos). Neste teste, um olho serve como alvo de tratamento e o outro como controle. As áreas em torno dos olhos, incluindo as pálpebras, conjuntiva, córnea e íris, também deverão ser examinadas durante o ensaio. Os olhos devem ser examinados por, pelo menos, 72 horas após a administração. O investigador deve concluir o teste com uma avaliação adequada das reações observadas. O tipo e extensão das investigações realizadas devem ser feitas de forma individual caso a caso. Um teste de tolerância ocular dose repetida (normalmente realizado em coelhos com administração diária durante quatro semanas). O desenho deste teste deve considerar os resultados da tolerância ocular dose única. O exame histológico deverá ser considerado caso a caso. Em ambos os experimentos, avaliações de diferentes tecidos em contato com o produto, bem como o corpo vítreo e do fundo ocular devem ser incluídos. A avaliação da tolerância ocular também é necessária para os produtos que não se destinam a serem administrados aos olhos, mas que se possa razoavelmente esperar que resultem em exposição no decurso da sua utilização clínica normal (por exemplo, loções ou gel utilizados para o tratamento da pele da face, xampu medicinal, etc.). Nestes casos, um teste de tolerância ocular usando uma única administração deverá ser executado. Via Retal: Modelo animal a ser utilizado: A seleção das espécies deve ser justificada, geralmente são utilizados coelhos ou cães. Dosagem: O volume de aplicação é baseado na dose terapêutica humana da formulação galênica ou no volume máximo aplicável à espécie animal. Administração: Normalmente uma ou duas vezes por dia, durante pelo menos 7 dias, de acordo com a utilização clínica. Parâmetros a serem Avaliados: 200
201 Observação da região anal e esfíncter anal, os sinais clínicos (por exemplo, sinais de dor) e fezes (por exemplo, sangue, muco). Eutanásia dos animais após a administração, necropsia e exame macroscópico da mucosa retal. Realização de exame histológico para diagnóstico, se necessário. Como nos medicamentos administrados sobre a pele, potencial de sensibilidade deve ser avaliado por meio de testes validados, geralmente realizados em cobaias. Via Vaginal: Modelo animal a ser utilizado: A seleção das espécies deve ser justificada, geralmente são utilizados coelhos, cães ou ratos. Dosagem: O volume de aplicação é baseado na dose terapêutica humana da formulação galênica ou no volume máximo aplicável à espécie animal. Administração: Normalmente uma ou duas vezes por dia, durante pelo menos 7 dias, de acordo com a utilização clínica. Parâmetros a serem Avaliados: Observação da região vaginal, sinais clínicos (por exemplo, sinais de dor) e secreção vaginal (por exemplo, sangue, muco). Se os animais forem submetidos à eutanásia após administração, realizar necropsia e exame macroscópico da mucosa vaginal e do sistema reprodutor. Realização de exame histológico para diagnóstico. Investigações adicionais (isto é, efeitos no muco cervical, ação espermicida) deverão ser consideradas caso a caso. Como nos medicamentos administrados sobre a pele, potencial de sensibilidade deve ser avaliado por meio de testes validados, geralmente realizados em cobaias. D. Potencial de Sensibilidade Sistemas de testes recomendados: Ensaio em cobaia: de acordo com o guia OECD para testes químicos nº406. Ensaio de Linfonodo Local: descrito na publicação: The Murine Local Lymph node assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds (NIHPublication Nº: ). Observações: A avaliação da Tolerância Local deve ser realizada antes de se iniciar a Fase 1 da Pesquisa Clínica e poderá ser parte de outros estudos de toxicidade. 7. Estudos de Carcinogenicidade Definição/Objetivo: Identificar substâncias que possam causar o desenvolvimento potencial de tumores em algum local por algum mecanismo, observando testes animais para o desenvolvimento de lesões como conseqüência da exposição, durante um tempo considerável de sua vida, por várias doses da substância teste e por uma via de administração apropriada. Estudos a serem realizados: O esquema padrão de carcinogenicidade compreende um estudo em roedor de longo prazo e: 1- Um estudo de curto e médio prazo em roedores in vivo que podem incluir modelos de iniciação/promoção em roedores, ou modelos de carcinogenicidade usando transgênicos ou roedores neonatais. Ou 2- Estudo a longo prazo de carcinogenicidade em uma segunda espécie roedora. Modelo animal a ser estudado: As espécies selecionadas devem ser apropriadas, baseadas nas seguintes considerações: a- Farmacologia b- Toxicidade em doses repetidas 201
202 c- Metabolismo d- Toxicocinética e- Via de administração Na ausência de evidências claras favorecendo uma espécie, é recomendado o uso do rato. Via de Administração: Os estudos devem ser conduzidos com administração pela mesma via de administração pretendida na clínica, quando possível. Caso haja demonstração de metabolismo e exposição sistêmica similares por outras vias de administração, então, os ensaios de carcinogenicidade podem ser conduzidos por uma única via, garantindo que órgãos importantes na via clínica sejam expostos adequadamente. A demonstração de exposição de tais órgãos pode ser evidenciada por dados farmacocinéticos. Dosagem: Alguns aspectos a serem considerados na escolha de doses são descritos a seguir, mas não limitados a: a) Toxicidade b) Farmacocinética c) Comparação da Área Sob a Curva em animais e seres humanos d) Saturação da absorção e) Farmacodinâmica f) Dose Máxima Possível g) Dose Limite h) Dose terapêutica humana i) Resposta Farmacodinâmica em humanos. Período de Observação: Para os estudos de longo prazo: 24 meses em ratos e, no mínimo, 18 meses em camundongos e hamsters. Parâmetros a serem avaliados: Os parâmetros deverão ser avaliados conforme orientação do Guia OECD 451. Todos os animais devem ser avaliados para morbidade ou mortalidade, geralmente no início e final de cada dia. Atenção especial deve ser dada ao desenvolvimento de tumores: o início, localização, dimensões, aparência, progressão de cada tumor macroscopicamente visível ou palpável. Devem ser verificados periodicamente sinais específicos de importância toxicológica além de peso corporal e consumo de ração/água. Hematologia: Esfregaços sanguíneos são realizados a critério do patologista. Necropsia: Os animais deverão ser submetidos à necropsia completa e detalhada, que inclui: um exame cuidadoso da superfície externa do corpo, dos orifícios e das cavidades craniana, torácica, abdominal e seus conteúdos. O peso dos órgãos não é normalmente parte de um estudo de carcinogênese. Os seguintes tecidos devem ser preservados em meio de fixação mais adequado para o tipo de tecido avaliado bem como para o exame histopatológico: todas as lesões macroscópicas, glândula adrenal, aorta, cérebro (incluindo seções do cérebro, cerebelo, e medula / ponte), ceco, colo do útero, glândula de coagulação, cólon, duodeno, epidídimo, olho (incluindo retina), a vesícula biliar (para outras espécies que não rato), glândula Harderiana, coração, íleo, jejuno, rins, glândula lacrimal (exorbital), fígado, pulmão, linfonodos (superficial e profunda), glândula mamária feminina, esôfago, pâncreas, ovário, glândula paratireóide, nervo periférico, hipófise, próstata, glândula salivar, vesícula seminal, músculo esquelético, pele, medula espinhal (em três níveis: cervical, médiotorácico e lombar), baço, estômago, testículos, timo, glândula tireóide, traquéia, bexiga, útero (incluindo colo do útero), vagina, e uma secção de medula óssea e / ou aspirado de medula óssea). Alguns achados podem sugerir a necessidade de examinar outros tecidos. Além disso, órgãos-alvo com base nas propriedades conhecidas da substância teste devem ser avaliados. 202
203 Observações: Necessidade da Realização de Ensaios de Carcinogenicidade - Casos de eventual necessidade de avaliação de potencial carcinogênico: (1) Estudos de carcinogenicidade devem ser realizados para quaisquer fármacos pretendidos para uso clínico contínuo de pelo menos 6 meses. Espera-se que muitos fármacos indicados para tratamento inferiores a 6 meses sejam utilizados de maneira contínua e intermitente. Em tais casos, como por exemplo, em terapias para rinite, depressão e ansiedade, há uma preocupação quanto ao potencial carcinogênico de tais fármacos. (2) Produtos da mesma classe de um fármaco cujo potencial carcinogênico foi demonstrado anteriormente. (3) Relação estrutura-atividade sugerindo potencial carcinogênico. (4) Evidência de lesões pré-neoplásicas em estudos de toxicidade em doses repetidas. (5) Retenção do composto ou seus metabólitos nos tecidos resultando em reações locais ou outras alterações patofisiológicas. (6) Quando houver preocupação com o potencial fotocarcinogênico, ensaios com aplicação dérmica (geralmente em camundongos) podem ser necessários. Casos onde ensaios de carcinogenicidade podem não ser necessários: (1) Quando a expectativa de vida da população-alvo for curta (menor que 2 a 3 anos). (2) Substâncias utilizadas topicamente (ex. via dérmica ou ocular) podem necessitar de ensaios de carcinogenicidade. Entretanto, fármacos que demonstrem pouca exposição sistêmica proveniente de uso tópico (dérmica ou ocular) em humanos podem não necessitar de ensaios orais para avaliar o potencial carcinogênico a órgãos internos. (3) Compostos comprovadamente genotóxicos, na ausência de outros dados, são presumidamente carcinógenos a seres humanos e não necessitam de estudos de longo prazo de carcinogenicidade. Entretanto, se o uso pretendido de tal fármaco for para terapias crônicas, um ensaio de toxicidade crônica (de até 1 ano) pode ser necessário para detectar lesões tumorais iniciais. (4) Substâncias endógenas administradas essencialmente como terapias repositórias (isto é, níveis fisiológicos) particularmente se houver experiência clínica com produtos similares (por exemplo, insulinas animais, hormônio do crescimento derivado da pituitária e calcitonina). (5) Substâncias utilizadas por um período curto de exposição ou de maneira infrequente (exemplo, anestésicos ou contrastes e agentes diagnósticos radio-marcados). Casos especiais devem ser considerados: (1) Peptídeos endógenos ou proteínas e seus análogos produzidos por síntese química, por extração/purificação de uma fonte animal/humana ou por métodos biotecnológicos como tecnologia de DNA recombinante podem requerer consideração especial. A condução de ensaios de carcinogenicidade pode ser importante nos casos de: a) produtos que demonstrem diferenças significativas nos efeitos biológicos quando comparados aos efeitos dos compostos naturais; b) produtos com modificações estruturais importantes; c) produtos cuja administração produza um aumento significativo em sua disponibilidade local ou sistêmica quando comparados aos produtos naturais em seres humanos. (2) Para diferentes sais, ácidos ou bases partes do mesmo grupo químico funcional, onde haja ensaios anteriores de carcinogenicidade, devem-se demonstrar evidências de que não haja diferenças significativas na farmacodinâmica, farmacocinética ou toxicidade. 203
204 Entretanto quando houver diferenças nos níveis de exposição e conseqüente toxicidade, então ensaios adicionais podem ser realizados para se determinar a necessidade ou não da realização de ensaios adicionais de carcinogenicidade. (3) Estudos de mecanismo de ação são úteis para avaliar a relevância dos achados tumorais nos animais, para a segurança humana. Testes adicionais podem ser necessários, quando houver necessidade da confirmação da presença ou ausência de potencial carcinogênico a seres humanos, investigando-se os mecanismos de ação. Momento de Submissão de estudos de carcinogenicidade Estudos de carcinogenicidade são recomendados em casos específicos. Caso tais estudos sejam necessários, estes deverão ser apresentados no momento do registro do produto para comercialização. Apenas em circunstâncias em que haja uma causa de preocupação de risco de carcinogenicidade, os resultados de tais estudos devem ser submetidos para suporte aos ensaios clínicos. Medicamentos desenvolvidos para tratar doenças graves podem ter seus estudos de carcinogenicidade concluídos após aprovação do registro. 8. Estudos de Interesse para a Avaliação da Segurança Farmacológica Definição/Objetivo Os estudos de interesse para a avaliação da segurança farmacológica são aqueles que pesquisam os potenciais efeitos farmacodinâmicos indesejáveis da substância teste nas funções fisiológicas dos diversos sistemas orgânicos em relação ao nível de exposição. A partir desses estudos são avaliadas funções vitais desenvolvidas pelos sistemas: nervoso central, cardiovascular e respiratório. Quando necessário, deve-se também avaliar o sistema urinário, nervoso autônomo, digestório, endócrino, imune e muscular esquelético. Via de Administração A mesma via a ser administrada em humanos, quando possível. Dosagem: São geralmente realizados com administração única. A duração deve ser baseada em critérios racionalmente elaborados: quando efeitos farmacodinâmicos ocorrem somente depois de certo período de tratamento, ou quando resultam de estudos não clínicos de doses-repetidas, ou quando resultados da utilização em humanos levam ao aumento de interesse sobre os efeitos na segurança farmacológica. Metabólitos: A avaliação dos metabólitos maiores é frequentemente obtida nos estudos em animais do composto principal. Se um ou mais metabólitos importantes para humanos estiverem ausentes ou forem produzidos em quantidades reduzidas nos animais, estudos específicos de segurança farmacológica para esse(s) metabólito(s) devem ser considerados. Estudos Realizados: A. Estudos para avaliar a segurança farmacológica da substância no Sistema Nervoso Central B. Estudos para avaliar a segurança farmacológica da substância no Sistema Respiratório C. Estudos para avaliar a segurança farmacológica da substância no Sistema Cardiovascular A. Estudos para avaliar a segurança farmacológica da substância teste no Sistema Nervoso Central: A.1. Modelo animal a ser utilizado: A escolha da espécie para a condução de estudos de segurança farmacológica do SNC deve ser justificada com base na suscetibilidade, sensibilidade, reprodutibilidade, disponibilidade de dados comparativos históricos, ou seja, de sua relevância para a obtenção de dados e a extrapolação das conclusões para seres humanos. 204
205 A.2. Parâmetros a serem avaliados: Efeitos da substância no sistema nervoso central devem ser avaliados apropriadamente. Observar: atividade motora, modificações comportamentais, coordenação, respostas reflexas sensório-motoras e temperatura corporal. Por exemplo, a bateria de observação funcional (FOB), o teste de Irwin modificado ou outro protocolo experimental apropriado podem ser utilizados. B. Estudos para avaliar a segurança farmacológica da substância no Sistema Respiratório: B.1. Parâmetros a serem avaliados: O sistema respiratório deve ser avaliado com base na freqüência respiratória e outras medidas de função respiratórias (como volume corrente ou saturação da hemoglobina). Para avaliação do sistema respiratório, observações clínicas não são geralmente adequadas, mas sim a quantificação dos parâmetros supracitados por metodologia adequada. C. Estudos para avaliar a segurança farmacológica da substância teste no Sistema Cardiovascular: Para avaliação do sistema cardiovascular devem ser estimadas: pressão sanguínea, freqüência cardíaca e eletrocardiograma. Avaliações in vivo, in vitro e/ou ex vivo, incluindo métodos para detecção de anormalidades de repolarização e condução devem também ser consideradas. Para avaliação do prolongamento da repolarização ventricular pode-se utilizar o ensaio IKR in vitro e o ensaio QT in vivo. C.1. Estudos eletrofisiológicos in vitro: Modelo animal a ser utilizado: Empregam-se preparação de células únicas (por exemplo: sistema de expressão heteróloga, cardiomiócitos desagregados) ou de múltiplas células (por exemplo: Fibras de Purkinje, músculos papilares, coração intacto). Preparações celulares e de tecidos para ensaios in vitro são obtidos de diferentes espécies animais incluindo: coelho, furão, cobaia, cão, suíno e ocasionalmente humano. O mecanismo iônico de repolarização em ratos e camundongos adultos difere de grande número de espécies, incluindo o homem. Portanto, a utilização de tecidos dessas espécies não é considerada adequada. C.2. Estudos eletrofisiológicos, in vivo: Modelo animal a ser utilizado: Cão, macaco, furão, suíno, coelho, cobaia. O mecanismo iônico de repolarização em ratos e camundongos adultos difere de grande número de espécies, incluindo o homem. Portanto, a utilização dessas espécies também não é considerada adequada nos testes in vivo. Parâmetros a serem Avaliados: O intervalo QT do Eletrocardiograma é o desfecho mais comumente utilizado para avaliar a substância teste em relação à repolarização ventricular. Outros parâmetros de interesse a serem avaliados podem incluir: pressão sanguínea, batimentos cardíacos, Intervalo PR, Duração QRS e arritmias. Observações: Quando a ocorrência de eventos adversos durante ensaios clínicos, suscitar preocupação para a segurança humana, informações adicionais deverão ser obtidas da literatura e/ou de novos estudos experimentais in vitro ou in vivo tais como os mencionados abaixo: Estudos Complementares: Sistema Nervoso Central: avaliações de farmacologia comportamental, aprendizado e memória, estudos de binding para ligantes específicos, neuroquímica, exames visuais, auditivos e/ou eletrofisiológicos. Sistema Cardiovascular: avaliações de débito cardíaco, contratilidade ventricular, resistência vascular, efeitos de substâncias endógenas e/ou exógenas sobre as respostas cardiovasculares. 205
206 Sistema Respiratório: avaliações de resistência e complacência das vias aéreas, pressão arterial pulmonar, ph sanguíneo, gasometria. Sistema Urinário: avaliações de volume urinário, gravidade específica, osmolalidade, ph, fluidos/balanço eletrolítico, proteínas, citologia, hemácias, bem como determinações químicas de uréia, creatinina, proteínas plasmáticas. Sistema Nervoso Autônomo: avaliações de ligação a receptores relevantes para o sistema nervoso autônomo, respostas funcionais para agonistas ou antagonistas in vivo ou in vitro, estimulação direta dos nervos autônomos e medição das respostas cardiovasculares e variabilidade do ritmo cardíaco. Sistema Digestório: avaliações de secreção gástrica, potencial de lesão gastrointestinal, secreção biliar, tempo de trânsito in vivo, contração ileal in vitro, ph gástrico. Outros Sistemas: avaliações de musculatura esquelética, funções imunes e endócrinas. Condições em que estudos de segurança farmacológica não são necessários: Testes de substâncias de uso tópico (por exemplo, cutânea ou ocular), onde a farmacologia da substância testada está bem caracterizada, e onde a exposição sistêmica ou a distribuição para outros órgãos ou tecidos seja baixa. Em casos de agentes citotóxicos administrados em pacientes vítimas de câncer em estágio terminal onde o benefício se sobressaia ao risco induzido pela substância teste, pode ser avaliado, pela Anvisa, a inicialização de pesquisas em seres humanos sem a devida conclusão desses estudos. Estudos adicionais de interesse para a avaliação da segurança farmacológica podem ser realizados durante a Pesquisa Clínica para esclarecer eventos adversos suspeitos ou observados durante os estudos não clínicos ou clínicos. No entanto, para agentes citotóxicos com novo mecanismo de ação, pode haver necessidade da condução de estudos de segurança farmacológica. Testes de produtos biotecnológicos que são altamente específicos para seu receptor alvo,muitas vezes é suficiente avaliar a segurança farmacológica como parte dos parâmetros de toxicidade e/ou estudos farmacodinâmicos; portanto, estudos de segurança farmacológica podem ser reduzidos ou suprimidos para estes produtos. Entretanto, para os produtos biotecnológicos que representam uma nova classe terapêutica e/ou os produtos não receptorespecíficos, uma avaliação mais extensa da segurança farmacológica deve ser considerada. Pode haver outras condições para as quais os estudos de segurança farmacológica sejam dispensáveis, por exemplo, novos sais com similaridades farmacocinéticas e farmacodinâmicas. 9. Estudos de Toxicocinética Definição/Objetivo: Toxicocinética é definida como a geração de dados farmacocinéticos tanto como um componente integral na condução de estudos de toxicidade não clínica como em estudos de suporte especificamente desenhados para avaliar a exposição sistêmica. Os dados obtidos desses estudos podem ser utilizados na interpretação de achados toxicológicos e na avaliação de sua relevância para a segurança clínica. A toxicocinética tem como objetivo primário a descrição da exposição sistêmica obtida em animais e a sua relação com o nível de dose e o tempo. Como objetivos secundários podem-se considerar: relato da exposição obtida em estudos de toxicidade para achados toxicológicos/contribuição para a avaliação da relevância desses achados para a segurança clínica, suporte à escolha de espécies e regimes de tratamento em estudos de toxicidade não clínica e o fornecimento de informações que em conjunto com achados toxicológicos contribuam para o desenho de estudos não clínicos de toxicidade subseqüentes. Ensaios de metabolismo in vitro e dados de ligação às proteínas plasmáticas em animais e humanos, além de dados de exposição sistêmica nas espécies utilizadas em ensaios de toxicidade de doses repetidas, devem ser, em geral, avaliados antes do início dos ensaios clínicos. Informações sobre farmacocinética (ex. absorção, distribuição, metabolismo e excreção), nas espécies testadas e informações bioquímicas in vitro relevantes às interações potenciais 206
207 da substância teste devem estar disponíveis antes da exposição a grande número de seres humanos ou tratamentos de longa duração (normalmente, antes da Fase III). A caracterização não clínica de metabólito(s) humano(s) deve(m) ser fornecida(s) somente quando tal(is) metabólito(s) for(em) observado(s) em exposição maior que 10% em relação ao total de exposição à substância e em níveis em humanos significativamente maiores que a máxima exposição observada nos estudos de toxicidade. Tais estudos devem ser conduzidos antes da Fase III. Alguns metabólitos não são toxicologicamente relevantes e sua caracterização não é requerida. A caracterização não clínica de metabólitos identificados como causa de preocupação toxicológica deve ser considerada caso a caso. Modelo animal a ser utilizado: Deve-se utilizar o modelo animal especificado para os estudos não clínicos de toxicidade e segurança correspondentes. O número de animais a ser utilizado deve ser o mínimo consistente com a produção adequada de dados de toxicocinética. Quando machos e fêmeas são utilizados no estudo, normalmente é estimada a exposição em animais de ambos os sexos, a menos que haja justificativa para não fazê-lo. Estudos de toxicocinética podem ser realizados tanto em todos como em uma parte representativa dos animais utilizados no estudo principal ou em grupos satélite. Em estudos com animais de grande porte as amostras para toxicocinética podem ser coletadas dos animais do estudo principal. Grupos satélite podem ser necessários para as espécies menores (roedores). O número de animais a ser utilizado deve ser o mínimo necessário para a geração adequada de dados toxicocinéticos. Dados toxicocinéticos não são necessários para estudos de diferentes durações, caso a posologia permaneça inalterada. Via de Administração: Similares àquelas preconizadas para uso humano, quando possível. Dosagem: O estabelecimento de níveis de dose nos ensaios de toxicidade é largamente direcionado por achados de toxicidade além das respostas farmacodinâmicas nas espécies testadas. Entretanto, os seguintes princípios toxicocinéticos podem contribuir para o estabelecimento de níveis de dose: Baixa: Nível de dose sem efeito tóxico. Média: Múltiplo apropriado da exposição de menor dose ou fração da maior dose. Alta: Normalmente determinada por considerações toxicológicas. Os tempos da coleta de amostra de fluidos corpóreos devem ser tão freqüentes quanto necessário, sem interferir com a condução normal do estudo ou causar estresse fisiológico desnecessário aos animais. Em cada estudo, o número de vezes da realização da coleta de amostras deve ser justificado demonstrando ser adequado para que se estime a exposição. A justificativa deve ser baseada nos dados cinéticos obtidos em estudos de toxicidade anteriores, estudos pilotos, estudos distintos de mesmo modelo animal, ou em outros modelos que possibilitem a extrapolação. Período de Observação: O período suficiente para avaliar a toxicocinética da substância, no modelo animal estudado. Parâmetros a serem avaliados: A quantificação da exposição pode ser representada por concentração plasmática (soro ou sangue) ou pela área sob a curva concentração da substância X tempo (AUC). Outras medidas, como por exemplo, excreções urinárias podem ser mais apropriadas para certas substâncias. Alguns parâmetros derivados, por exemplo, biodisponibilidade, meia-vida, volume de distribuição da substância teste, podem ser importantes na interpretação dos dados de toxicocinética. Portanto, a seleção dos parâmetros e tempo de coleta deve ser feita, caso a caso, considerando-se os princípios observados neste Guia. Observações: 207
208 O desenvolvimento de um medicamento é um processo dinâmico que envolve intercâmbio de informações obtidas em estudos clínicos e não clínicos. Por isso, não se recomenda a adoção de procedimentos rígidos e pormenorizados para a submissão de estudos toxicocinéticos. A coleta de dados toxicocinéticos pode não ser necessária em todos os estudos e o julgamento científico irá indicar os casos em que esses dados podem ser úteis. A necessidade de obter dados toxicocinéticos e de avaliar a exposição em cada estudo individual de toxicidade deve ser baseada em uma abordagem flexível e estabelecida de forma individualizada, garantindo-se a produção de informações suficientes para avaliar os riscos e a segurança da exposição de seres humanos. 1- Seguem abaixo considerações sobre a análise toxicocinética a ser aplicada nos diversos estudos toxicológicos: Estudos de Toxicidade de Dose Única: Estudos toxicocinéticos, se necessários, podem ser realizados após a finalização dos estudos de toxicidade de dose única para responder questões específicas que surgiram durante o estudo. Resultados dos estudos cinéticos de dose única podem auxiliar na escolha da formulação e na predição da medida e duração da exposição durante o intervalo de dose. Isto poderá auxiliar na seleção apropriada dos níveis de doses a serem utilizados em estudos posteriores. Esses estudos muitas vezes são realizados em uma fase muito inicial de desenvolvimento, antes que um método bioanalítico tenha sido desenvolvido, portanto o monitoramento toxicocinético desses estudos, normalmente não é possível. As amostras de plasma em tais estudos podem ser colhidas e armazenadas para análise posterior, se necessário. Os dados adequados da estabilidade do analito na matriz amostrada seriam então necessários. Alternativamente, estudos toxicocinéticos adicionais podem ser realizados após a conclusão de um estudo de toxicidade de dose única, a fim de responder a perguntas específicas que podem surgir. Estudos de Toxicidade de Doses Repetidas: Para esses estudos, o regime de tratamento e as espécies a serem utilizadas devem ser selecionados considerando-se os princípios da farmacocinética e da farmacodinâmica. Isso pode não ser possível nos estudos iniciais quando dados farmacocinéticos provenientes de animais ou de seres humanos ainda não estão disponíveis. A toxicocinética deve ser incorporada apropriadamente no desenho desses estudos. Estudos de Genotoxicidade: Em casos de resultados negativos de estudos de genotoxicidade in vivo, pode ser necessária a demonstração da exposição sistêmica na espécie utilizada ou a caracterização da exposição em tecido indicado. Estudos de Carcinogenicidade: É necessária a estimativa da exposição sistêmica para que haja a confirmação de que seja possível extrapolar dados do modelo animal para o modelo humano. O regime de tratamento e a espécie a ser selecionada devem, tanto quanto possível, ser determinados considerando as informações farmacocinéticas e toxicocinéticas disponíveis. Estudos de Toxicidade Reprodutiva: É preferível ter algumas informações da farmacocinética antes de se iniciar os estudos de toxicidade reprodutiva, uma vez que, essas podem sugerir a necessidade de ajustes na escolha da espécie, no desenho do estudo e na dose determinada. Para estudos com fêmeas prenhes e lactantes, o regime de tratamento durante a exposição deve ser selecionado com base em achados toxicológicos, princípios farmacocinéticos e toxicocinéticos. Deve-se considerar o fato de que a cinética poderá diferir entre fêmeas prenhes e não prenhes. As avaliações toxicocinéticas podem envolver exposições de embriões, fetos ou recém-nascidos. A secreção da substância no leite pode ser avaliada para definir o papel da exposição em recém nascidos. Em algumas situações, estudos adicionais podem ser necessários ou apropriados para avaliar a transferência embrio/fetal e a secreção da substância no leite. 208
209 2-Para análise toxicocinética devem-se considerar fatores como: ligação às proteínas, propriedades receptoras e perfil metabólico. 3-Em alguns casos pode ser necessária a avaliação de metabólitos como, por exemplo: -Quando a substância é uma pró-substância teste, -Quando a metabolização da substância resulta em um ou mais metabólitos toxicologicamente ou farmacologicamente ativos, -Quando a substância é extensamente metabolizada. 4-Parâmetros cinéticos, como por exemplo, níveis de pico plasmático, AUC (área sob a curva concentração da substância X tempo), e informações sobre a Máxima Dose Tolerada (MTD) em animais utilizados para estudos préclínicos, auxiliam na definição de doses a serem administradas durante a Fase 1 da Pesquisa Clínica. 10. Estudos não clínicos de segurança para o desenvolvimento e registro de ADF (Associação em Dose Fixa) de medicamentos sintéticos e semissintéticos de uso oral. Durante o desenvolvimento de uma associação, os estudos não clínicos possuem o objetivo de caracterizar o efeito do uso combinado dos ativos do ponto de vista farmacológico, farmacocinético e toxicológico, identificando efeitos aditivos, sinérgicos ou antagônicos da associação. A necessidade e a extensão de estudos não clínicos com a associação devem ser definidas com base nas características individuais dos princípios ativos, no potencial de interações entre esses ativos, nos estudos não clínicos e clínicos já existentes tanto para associação quanto para as monodrogas e no objetivo terapêutico dessa associação. Uma avaliação desse conjunto de fatores é necessária para o delineamento do desenvolvimento não clínico do produto ou para se concluir que a realização de estudos não clínicos com a associação não é necessária. Independente da avaliação, a realização de estudos não clínicos de segurança é necessária quando a formulação possuir um ou mais excipientes cuja segurança não esteja bem estabelecida ou quando o perfil de impurezas da associação é significativamente diferente dos produtos contendo as monodrogas NOVAS ASSOCIAÇÕES CONTENDO MONODROGAS JÁ REGISTRADAS Considerações gerais As considerações que serão feitas neste item partem do pressuposto que cada princípio ativo separadamente apresenta sua eficácia e segurança bem estabelecida. Se houverem preocupações específicas com relação à segurança das monodrogas ou lacunas com relação aos estudos não clínicos das mesmas, estudos adicionais poderão ser necessários. É necessário que se faça uma avaliação dos medicamentos que estão registrados frente à associação em desenvolvimento, verificando se os dados clínicos e não clínicos já disponíveis são suficientes para subsidiar a indicação, dose terapêutica, posologia, duração do tratamento e população alvo, propostos para a associação. Por exemplo, medicamentos registrados para uso agudo podem não possuir estudos não clínicos para subsidiar uma indicação de uso crônico. Quando os dados existentes para as monodrogas não forem condizentes com as características da associação proposta, poderá ser necessária a realização de estudos não clínicos além dos aqui descritos. Para associações constituídas de princípios ativos já registrados para os quais já existam evidências suficientes para comprovação da eficácia e segurança da associação, estudos de segurança com a combinação em animais geralmente não são necessários. Para as demais associações as considerações sobre os estudos não clínicos serão discutidas nos itens seguintes Avaliação da segurança da associação 209
210 Se os dados existentes para cada monodroga separadamente forem condizentes com a indicação, dose terapêutica, posologia, duração do tratamento e população alvo pleiteados para associação, sugere-se que alguns fatores relevantes para a avaliação da segurança sejam considerados para identificar se são necessários estudos não clínicos adicionais para a associação e quais estudos não clínicos precisarão ser conduzidos. A avaliação com base nesses itens permite identificar possíveis preocupações clínicas com relação à segurança da associação que suscitem a necessidade de estudos não clínicos adicionais. a) Informações disponíveis sobre o uso da associação em humanos: Estes dados podem ser suficientes para a avaliação da segurança da associação ou apontar pontos que podem ser motivo de preocupação clínica e precisem ser mais bem estudados. b) Possibilidade de interações farmacodinâmicas: As monodrogas podem ter afinidade pelos mesmos receptores biológicos ou produzirem efeitos fisiológicos semelhantes, relacionados ou não ao seu mecanismo de ação. c) Possibilidade de interações farmacocinéticas: Uma monodroga pode alterar a absorção ou excreção da outra, mudar sua distribuição em um ou mais tecidos, ou alterar sua taxa de metabolização. As monodrogas podem competir entre si pela ligação às proteínas plasmáticas, aumentando assim a fração livre no plasma de uma delas e sua distribuição para os tecidos. d) Possibilidade de interações toxicológicas: Por exemplo, princípios ativos que possuem toxicidade no mesmo órgão alvo. Nesses casos, a toxicidade da associação no órgão alvo pode ser mais severa, e as doses estabelecidas como não tóxicas para as monodrogas podem não se aplicar para o uso combinado. e) Margem de segurança para cada monodroga da associação: Se um ou mais princípios ativos possuírem faixa terapêutica estreita, a possibilidade de interações farmacocinéticas possui especial importância na avaliação da segurança da associação, principalmente se o efeito tóxico dos ativos for irreversível ou não puder ser monitorado clinicamente. f) Possibilidade competição entre as monodrogas, alteração na atividade ou níveis plasmáticos de uma mesma enzima ou outras moléculas intracelulares. g) Possibilidade de interações químicas: Uma molécula pode modificar quimicamente outra (através de oxidação, metilação, etc) levando ao surgimento de uma nova entidade molecular. h) Possibilidade de um fármaco comprometer a efetividade do outro, principalmente em indicações para doenças graves com risco de morte Estudos de Toxicidade Geral Para associações constituídas de princípios ativos registrados para os quais já exista adequada experiência clínica do uso combinado em humanos, os estudos de toxicidade geralmente não são necessários para subsidiar os estudos clínicos que serão conduzidos com a associação ou o registro do medicamento, desde que não haja nenhuma preocupação com relação à toxicidade da associação mediante avaliação de acordo com o item (por exemplo, toxicidade sob o mesmo órgão alvo) ou outro motivo que represente grave preocupação clínica (por exemplo, um dos componentes da formulação pertencer a uma classe terapêutica que esteja associada a um tipo específico de toxicidade). A margem de segurança das monodrogas e a capacidade de se monitorar os eventos adversos relacionados à toxicidade em humanos devem ser consideradas na decisão sobre a condução ou não dos estudos. Quando conduzidos para avaliar efeitos toxicológicos específicos, os estudos não clínicos devem ser feitos antes do início dos estudos clínicos com a associação. Para associações de duas ou mais moléculas já registradas para as quais não haja adequada experiência clínica do uso combinado é necessária à realização de estudos não clínicos de toxicidade com a associação, tais estudos são necessários para subsidiar os estudos clínicos de larga escala e longa duração assim como o registro. Para avaliação 210
211 da toxicidade da associação, um estudo ponte pode ser apropriado desde que tenha duração suficiente para esclarecer a preocupação com relação à toxicidade da associação. Para subsidiar os estudos clínicos com a associação, os estudos de toxicidade devem ter duração equivalente à pretendida para os estudos clínicos, tendo um limite máximo de 90 dias. Para o registro, estudos com 90 dias de duração podem ser considerados para indicação de uso crônico. A necessidade de estudos com maior duração irá depender dos efeitos observados no estudo da combinação de fármacos quando comparados aos induzidos pelas monodrogas. Dependendo do tempo de tratamento que será proposto para a associação, estudos com tempo de duração menor poderão ser suficientes para subsidiar o registro. Geralmente, os estudos ponte podem ser realizados em apenas uma espécie animal relevante. A escolha do modelo mais relevante deve ser cientificamente justificada com base em aspectos farmacodinâmicos, farmacocinéticos, de metabolismo, órgãos alvos e sensibilidade toxicológica. Testes adicionais em outras espécies podem ser necessários caso um efeito tóxico não esperado seja identificado. O desenho dos estudos não clínicos com a associação irá depender das características farmacológicas, toxicológicas e farmacocinéticas das monodrogas individualmente e das indicações pleiteadas, população alvo pretendida e outros dados disponíveis para a associação. Devem ser delineados de maneira que se possa fazer uma comparação dos dados obtidos para a associação com os dados das monodrogas. A inclusão de braços comparadores com altas doses das monodrogas pode ser considerada Estudos de Genotoxicidade Os estudos de genotoxicidade para a associação geralmente não são necessários se os componentes individuais da formulação tiverem sido adequadamente testados com relação a esse aspecto. Quando algum dos princípios ativos da associação possui potencial genotóxico, estudos adicionais para avaliar a possibilidade de potencialização desse efeito podem ser necessários. Uma avaliação caso a caso com base nas informações disponíveis para cada monodroga deve ser feita para avaliar como a possibilidade de aumento da genotixicidade pode influenciar no balanço risco/benefício do uso da associação para a indicação pleiteada Estudos de Carcinogenicidade Os estudos de carcinogenicidade para a associação geralmente não são necessários se os componentes individuais da formulação tiverem sido adequadamente testados com relação a esse aspecto. O potencial de carcinogenicidade da associação pode ser avaliado com base nas informações disponíveis para as monodrogas. Quando algum dos princípios ativos da associação possuir potencial carcinogênico, estudos adicionais para avaliar a possibilidade de potencialização desse efeito podem ser necessários. Uma abordagem pode ser a inclusão nos estudos de toxicidade repetida de desfechos relevantes para a avaliação de carcinogenicidade, para que se possa avaliar qualquer preocupação clínica com relação a esse ponto. Os estudos de carcinogenicidade são recomendados para associações de indicação para uso crônico quando nos achados não clínicos for observada incidência estatisticamente significativa de lesões pré-neoplásicas em órgãos ou tecidos Estudos de Toxicidade Reprodutiva Os estudos de toxicidade reprodutiva para a associação geralmente não são necessários se os componentes individuais da formulação tiverem sido adequadamente testados com relação a esse aspecto. A decisão pela necessidade ou não de estudos de toxicidade reprodutiva irá depender das propriedades de cada ativo e do potencial de interação entre eles. Nos casos em que a população alvo da associação incluir mulheres em idade fértil e os estudos com as monodrogas já tiverem apontado risco embrio-fetal, estudos com a associação não serão necessários apenas se o risco para o 211
212 desenvolvimento humano já tiver sido identificado. Se os estudos de desenvolvimento embrio-fetal das monodrogas não tiverem demonstrado nenhum risco ao desenvolvimento humano para nenhum dos fármacos individuais, os estudos não clínicos com a associação somente precisam ser feitos se houver alguma suspeita com base nas propriedades de cada fármaco de que há um risco do uso da combinação para humanos. Quando necessário, o estudo pode ser conduzido na espécie mais apropriada, baseando-se no conhecimento prévio que se tem dos fármacos. Caso um risco significativo seja identificado somente em um trimestre da gravidez os estudos devem avaliar os efeitos da associação nos outros trimestres da gravidez também Estudos de Segurança Farmacológica Os estudos de segurança farmacológica para a associação geralmente não são necessários se os componentes individuais da formulação tiverem sido adequadamente testados com relação a esse aspecto. A necessidade de condução de estudos de segurança farmacológica com a associação irá depender dos tipos de interações que podem ocorrer entre os princípios ativos. Os estudos podem ser necessários quando os princípios ativos que constituirão a associação possuem toxicidade no mesmo órgão alvo ou sistema (por exemplo: cardíaco, respiratório, SNC, etc), quando um ou mais ativos pertencem a classes terapêuticas associadas com algum tipo específico de toxicidade (por exemplo: prolongamento do intervalo QT), quando a indicação inclui população com algum tipo de comprometimento (por exemplo: pacientes com insuficiência renal) ou quando qualquer outra preocupação clínica for identificada Novas associações contendo uma ou mais moléculas novas no país Nos casos em que um ou mais princípios ativos da associação forem novas moléculas no país, os estudos não clínicos devem seguir as mesmas diretrizes de desenvolvimento e registro de um novo medicamento. 212
213 Guia para Registro de Novas Associações em Dose Fixa INTRODUÇÃO Associações medicamentosas podem ser utilizadas em diferentes condições de saúde, e devem ter qualidade, segurança e eficácia comprovadas, com riscos e benefícios bem definidos. O registro de uma associação medicamentosa se justifica quando há uma população de pacientes definida para a qual uma combinação particular de ativos em uma razão fixa de doses tenha demonstrado ser segura e eficaz, e quando todos os princípios ativos contribuem para o efeito terapêutico. O presente Guia é uma orientação para o Registro de Novas Associações em Dose Fixa de Medicamentos Sintéticos e Semi-Sintéticos, para uso oral, conforme legislação específica. O objetivo deste Guia é esclarecer os requisitos regulatórios de eficácia e segurança necessários para o registro de uma ADF na Anvisa. São abordados neste Guia somente os requisitos para os estudos não clínicos e clínicos, sendo que todos os demais itens das normas específicas devem ser cumpridos para o registro sanitário do produto. Para efeito deste Guia, foram adotadas as seguintes definições: Associação em dose fixa: uma combinação de dois ou mais princípios ativos em uma razão fixa de doses em uma mesma forma farmacêutica. O termo foi utilizado para designar o produto em sua formulação final, exatamente como será registrado. Associação: uma combinação de princípios ativos em uma determinada associação de doses. O termo foi utilizado de maneira genérica, para designar uma associação específica de princípios ativos independentemente da formulação, ou seja, os princípios ativos isolados administrados concomitantemente ou os princípios ativos em uma mesma forma farmacêutica. Aditismo: o efeito combinado de fármacos A e B é igual à soma aritmética dos efeitos dos fármacos individuais, A + B, nas doses escolhidas. Sinergismo: a ação simultânea de dois fármacos resulta em um efeito superior ao que podia ser esperado com a simples adição dos efeitos individuais dos fármacos. Capítulo 1 CONSIDERAÇÕES GERAIS Para o registro de novas associações são aceitos no máximo 3 (três) princípios ativos na mesma formulação por apresentação oral, ou quatro, caso um deles seja a cafeína. Os dados de eficácia e segurança de uma associação podem ser obtidos a partir de estudos clínicos realizados com medicamentos contendo os princípios ativos isolados administrados concomitante, ou com a associação em dose fixa desses princípios ativos. Não são aceitos, para fins de registro de uma associação, a apresentação exclusiva de estudos clínicos nos quais foram utilizados os princípios ativos de forma isolada, ou seja, estudos nos quais não foi avaliado o uso concomitante dos princípios ativos. Quando os dados clínicos apresentados forem dos medicamentos com os princípios ativos isolados com administração concomitante, estudos de bioequivalência entre as monodrogas administradas concomitantemente e a ADF são necessários para que os dados dos estudos clínicos sejam extrapolados para a ADF que se pretende registrar. Todo suposto benefício de uma associação deve ser confirmado por meio de estudos clínicos. Os riscos e benefícios não podem ser baseados em considerações somente teóricas ou extrapolados de outros dados. Os estudos clínicos apresentados para obtenção do registro devem ser relativos às concentrações, regime posológico, indicações terapêuticas, população alvo, dentre outros aspectos pretendidos. 213
214 A origem dos dados de eficácia e segurança de uma associação pode variar de acordo com o nível de evidência disponível na literatura. As evidências não clínicas e clínicas citadas neste Guia podem ser constituídas de dados originais do desenvolvimento da associação ou dados da literatura ou ambos. Quando os dados clínicos forem provenientes da literatura científica, os artigos apresentados devem estar publicados em revistas indexadas e cumprir com os requisitos estabelecidos no item 6.3 deste Guia. Nesses casos, estudos de bioequivalência são necessários para que os dados da literatura científica apresentados possam ser extrapolados para a ADF que se pretende registrar. Capítulo 2 SITUAÇÕES PARA O REGISTRO DE UMA ADF A solicitação de Registro de Nova Associação em Dose Fixa no país pode se enquadrar em uma das seguintes situações: Situação 01: a ADF possui os mesmos princípios ativos, nas mesmas concentrações e posologia que um regime de tratamento com uso concomitante das monodrogas que possuem seu perfil de segurança e eficácia estabelecido por meio de evidência científica disponível na literatura. Nesse caso, poderá ser apresentado relatório clínico baseado em dados de literatura publicados em revistas indexadas, desde que atendidos os requisitos estabelecidos no item 6.3 deste Guia. Situação 02: as monodrogas registradas possuem perfil de eficácia e segurança bem estabelecido, mas a associação ainda não foi estudada nas doses e para as indicações terapêuticas que se pretende pleitear, ou seu perfil de eficácia e segurança não está estabelecido por meio de evidência científica disponível na literatura. Nesse caso, sempre será necessária a condução de estudo clínico Fase III, e eventualmente de estudos clínicos Fase II com a associação. Situação 03: a ADF proposta possui um ou mais princípios ativos novos no país. Nesse caso, sempre será necessária a condução de estudos não clínicos e estudos clínicos Fase I, II e III com o produto que se pretende registrar. Os estudos não clínicos, clínicos e de biodisponibilidade relativa necessários, poderão variar de acordo com a situação na qual a ADF se enquadra. De acordo com a classe terapêutica da ADF, a Anvisa poderá editar, em conjunto com Sociedades Médicas Especializadas, Guias de orientação científica para o registro sanitário desses produtos, citando as associações com maior potencial de enquadramento nas situações 1 e 2. Capítulo 3 JUSTIFICATIVA DA RACIONALIDADE DA ADF A justificativa da racionalidade da associação pode ser entendida como uma explicação sobre o racional de desenvolvimento do produto, que deve incluir considerações acerca das questões farmacocinéticas, farmacodinâmicas e terapêuticas do uso concomitante dos princípios ativos. As questões farmacocinéticas devem ser descritas conforme o tipo de associação. Quando uma das justificativas da racionalidade da associação é a ocorrência de interação farmacocinética, como nos casos em que um ativo aumenta a biodisponibilidade do outro, essa interação deve estar bem descrita, e os possíveis riscos e benefícios dessa interação devem ser adequadamente demonstrados. Dentre as questões terapêuticas, deve-se observar que uma associação se justifica quando existe uma população definida que será beneficiada pela terapia combinada. Nos casos em que a justificativa da associação for a ocorrência de comorbidade, dados epidemiológicos da coexistência das doenças na população devem ser apresentados para justificar a racionalidade da associação. O mesmo raciocínio se aplica quando o objetivo da associação é aliviar diferentes sintomas de uma doença. É necessário que tais sintomas ocorram de forma simultânea, em intensidade clinicamente relevante e por um período de tempo para o qual o tratamento concomitante seja apropriado. 214
215 Deve-se considerar também, a compatibilidade entre o regime posológico e recomendações de administração, aprovados para os tratamentos com as monodrogas. Por exemplo, se a freqüência de doses ao longo do dia é a mesma e se ambas possuem a mesma recomendação de administração. Na justificativa da racionalidade da associação, é necessário que se faça uma argumentação sobre suas vantagens frente às desvantagens. Todas as possíveis vantagens e desvantagens devem ser listadas e discutidas de acordo com as evidências científicas disponíveis. Dentre as possíveis vantagens de uma associação, pode-se citar: efeito aditivo ou sinérgico dos princípios ativos associados para cada indicação terapêutica ou; maior eficácia dos princípios ativos associados, sem aumento dos riscos quando comparado às monodrogas ou; mesma eficácia terapêutica com redução dos eventos adversos quando comparado às monodrogas ou; redução do desenvolvimento de resistência microbiana (para antimicrobianos) ou; simplificação do regime terapêutico. Dentre as possíveis desvantagens de uma associação, pode-se citar: dificuldade de ajuste individual da posologia para diferentes pacientes. Isso pode ocorrer quando qualquer uma das monodrogas possui esquema de escalonamento de dose para adequação do tratamento do paciente, ou quando são necessários ajustes de doses para populações especiais ou; perfil de eventos adversos com a associação diferente daquele observado com princípios ativos utilizados separadamente, como aparecimento de um evento adverso ainda não relatado para a monodroga ou aumento da ocorrência de eventos adversos já descritos para os princípios ativos isolados. Quando as monodrogas registradas possuem esquema de escalonamento de dose ou diferentes esquemas posológicos, a escolha de cada combinação de dose da ADF deve ser justificada quanto a relevância clínica. Nos casos de necessidade de ajuste de dose para populações especiais, essa informação deve estar claramente descrita em texto de bula. Capítulo 4 ESTUDOS NÃO CLÍNICOS A necessidade de estudos não clínicos é definida de acordo com as evidências científicas disponíveis para a associação. Os estudos não clínicos serão dispensados quando a associação de princípios ativos já tenha sido extensivamente utilizada em humanos, na faixa terapêutica que se pretende registrar, por um longo período e, possuir seu perfil de segurança estabelecido. No processo de registro, nos casos em que não forem apresentados ensaios não clínicos, o solicitante deve enviar a justificativa técnica para a não realização dos estudos, citando as referências bibliográficas que embasam a justificativa. Os estudos não clínicos serão necessários quando: a formulação possuir excipientes cuja segurança não esteja bem estabelecida ou quando o perfil de impurezas da associação é significativamente diferente dos produtos contendo as monodrogas; o perfil de segurança da associação ainda não estiver bem estabelecido; a faixa terapêutica proposta ainda não tiver sido estudada tanto para os princípios ativos isolados, como para os associados; entre os princípios ativos da associação, um ou mais forem novos no país. Nos casos em que um ou mais princípios ativos da associação forem novas moléculas no país, os estudos não clínicos devem seguir os mesmos parâmetros de um registro de Medicamento Novo. 215
216 Quando, o perfil de segurança da associação ainda não está estabelecido, estudos não clínicos devem ser conduzidos para investigar possíveis efeitos toxicológicos aditivos ou sinérgicos. Os estudos não clínicos, quando necessários, devem ser conduzidos de acordo com o Guia para a Condução de Estudos Não Clínicos de Segurança Necessários ao Desenvolvimento de Medicamentos da Anvisa. Capítulo 5 ESTUDOS DE BIODISPONIBILIDADE RELATIVA Os estudos de biodisponibilidade podem ter diferentes objetivos dependendo da situação em que a ADF que se pretende registrar se enquadra. O estudo pode ser delineado com a finalidade de estudar as interações farmacocinéticas entre os princípios ativos presentes na associação ou com a finalidade de estabelecer a bioequivalência entre o produto que será registrado e os produtos que foram utilizados nos estudos clínicos que subsidiam o registro. A possibilidade de interações farmacocinéticas entre os princípios ativos deve sempre ser considerada. O solicitante do registro deve fornecer dados que demonstrem que essas interações não ocorrem ou, se ocorrem, que estão claramente definidas. Nas situações 02 e 03, as possíveis interações farmacocinéticas entre os princípios ativos ainda não são conhecidas ou não estão bem estabelecidas. Nesses casos, estudos de biodisponibilidade relativa devem ser conduzidos para elucidar quaisquer interações farmacocinéticas entre os princípios ativos. Esses estudos são obrigatórios e devem preceder os estudos clínicos que serão conduzidos com a associação. Os estudos de bioequivalencia são sempre necessários quando os estudos clínicos que serão apresentados para o registro da associação não foram realizados com a ADF que será registrada. Os medicamentos que serão utilizados como referência para estes estudos devem pertencer a Lista de Medicamentos de Referência da Anvisa. Os estudos de bioequivalencia se aplicam à situação 01, ou seja, quando a nova ADF possui os mesmos princípios ativos, nas mesmas concentrações e mesma posologia que um regime de tratamento com uso concomitante das monodrogas que possuem seu perfil de segurança e eficácia bem estabelecidos por meio de evidência científica disponível na literatura, e as interações farmacocinéticas já estão bem definidas. Normalmente, os estudos clínicos disponíveis apresentados para o registro são conduzidos com as monodrogas registradas administradas concomitantemente. Nesse caso, para a aprovação da nova ADF, é necessário determinar se a velocidade e extensão da absorção de cada um dos componentes combinados é a mesma após administração concomitante destes componentes isolados, podendo assim, os dados de eficácia e segurança disponíveis para a associação serem extrapolados para a ADF que se pretende registrar. Caso durante a avaliação do registro se conclua que os dados clínicos apresentados de uma associação são insuficientes para a comprovação de eficácia e segurança, o estudo de bioequivalencia não pode ser considerado isoladamente, como evidência suficiente para o registro do produto. Nas situações 2 e 3 os estudos de bioequivalência também podem ser necessários, quando nos estudos clínicos tenham sido estudadas as monodrogas administradas concomitante, e não a ADF que será registrada. No caso da situação 03, além das considerações sobre as interações farmacocinéticas entre os ativos da ADF, todos os estudos para a nova molécula deverão seguir o que está determinado para o registro de Medicamento Novo. Os estudos de biodisponibilidade relativa deverão seguir as normas e Guias vigentes da Anvisa que regulamentam o tema. ESTUDOS CLÍNICOS 6.1 Requisitos Gerais Capítulo 6 216
217 Os dados de eficácia e segurança de uma associação podem ser obtidos a partir de estudos clínicos realizados com a administração concomitante de medicamentos com os princípios ativos isolados ou com a ADF que se pretende registrar. Os estudos clínicos que subsidiarão o registro podem ser provenientes da literatura (situação 01) de estudos originais desenvolvidos pelo detentor do produto ou de ambos. Independentemente da origem, os estudos clínicos que serão apresentados devem estar de acordo com os requisitos descritos neste Capítulo. Em cada caso deve-se avaliar a necessidade de estudos de biodisponibilidade relativa conforme Capítulo 5 deste Guia. Para o registro de uma nova associação, independentemente de se enquadrar na situação 01, 02 ou 03, devem ser apresentados estudos clínicos Fase III, randomizados, controlados, referentes à associação para cada indicação terapêutica. Os dados devem demonstrar que cada ativo contribui para o efeito da associação ou, nos casos em que um dos ativos potencializa o efeito do outro, essa superioridade da associação deve ser demonstrada. Os medicamentos que são utilizados como comparadores nos estudos clínicos obrigatoriamente devem ser produtos já registrados que possuem qualidade, eficácia e segurança já conhecidos. Os estudos clínicos devem contemplar as doses e posologia que se pretende registrar. Nos casos em que diferentes razões de dose dos princípios ativos serão objeto de registro, é importante que os dados clínicos permitam avaliar o perfil de risco e benefício de cada uma delas. Para as associações em cujos estudos farmacocinéticos fique demonstrada a existência de interações farmacocinéticas, os estudos clínicos de eficácia e segurança devem avaliar a influência dessa interação no perfil de eficácia e segurança da associação. O desenho e os desfechos escolhidos para os estudos devem sempre ser justificados e irão variar de acordo com o racional terapêutico da associação e com as indicações terapêuticas pretendidas. Por exemplo, quando o objetivo do estudo é demonstrar eficácia superior da associação em comparação ao tratamento com as monoterapias, um ensaio de superioridade pode fornecer a confiança estatística necessária para a comprovação dessa hipótese. Nos casos em que a hipótese é diminuição dos eventos adversos com manutenção da eficácia, delineamentos diferentes podem ser definidos para cada desfecho que será estudado: não inferioridade para o desfecho de eficácia e superioridade para o desfecho de segurança. Os dossiês devem trazer uma discussão tanto da significância estatística como da relevância clínica dos resultados. Somente serão aprovadas as indicações, doses e posologias utilizadas nos estudos clínicos para a população estudada. Na submissão da solicitação de registro não é necessário enviar estudos clínicos das monodrogas. Todos os dados clínicos devem ser da associação que será registrada, inclusive, os dados de Farmacovigilância que possam vir a ser apresentados. Após registro do produto, no caso de existência de qualquer problema de segurança relacionado a um dos princípios ativos que compõem a ADF, essa estará submetida às mesmas medidas de segurança tomadas pela Anvisa para o princípio ativo em questão. 6.2 Requisitos Específicos Para as associações que se enquadram na situação 01, o relatório clínico poderá ser baseado em dados de literatura da associação publicados em revistas indexadas conforme item 6.3, devendo-se em todos os casos, apresentar publicações referentes a estudos clínicos Fase III, randomizados, controlados, referentes à ADF ou ao uso concomitante dos princípios ativos que a compõem. Devem-se observar as exigências relativas aos estudos de bioequivalência descritos no Capítulo 5. Os seguintes aspectos devem ser estabelecidos e comprovados por meio de estudos clínicos: indicações terapêuticas propostas (com especificação de uso em primeira ou segunda linha quando for o caso); população alvo; posologia proposta; possíveis interações medicamentosas; dados de segurança; manutenção do perfil de risco e benefício da 217
218 ADF em relação ao uso dos princípios ativos isolados. No caso de melhora de adesão fazer parte do racional para registro da ADF, tal aspecto deve ser documentado fundamentado em dados científicos. Para as associações que se enquadram na situação 02, podem ser necessários estudos clínicos de fase II, para estabelecimento das doses ideais que serão utilizadas nos estudos de fase III. Por exemplo, nos casos em que um dos princípios ativos potencializa o efeito dos outros, combinações de concentrações diferentes dos ativos precisam ser testadas e dados de dose resposta devem ser apresentados para justificar as combinações de dose escolhidas para a ADF. Caso esses estudos não sejam conduzidos, será necessário apresentar justificativa com base em evidências científicas para a determinação das doses dos estudos de fase III. Para as associações que se enquadram na situação 3, os estudos clínicos devem cumprir os mesmos requisitos exigidos para registro de Medicamento Novo. Nesse caso, o produto será classificado como medicamento novo e deverá atender aos requisitos legais para este registro. 6.3 Dados de literatura A comprovação de eficácia e segurança com base em dados de literatura deve ser feita por meio de estudos clínicos publicados em revistas indexadas. Os medicamentos utilizados nos estudos clínicos devem possuir qualidade, eficácia e segurança já conhecidos e um histórico amplo de comercialização. A avaliação dos estudos contemplará a qualidade e consistência da literatura apresentada, além da sua adequação para o registro que está sendo proposto com relação a via de administração, forma farmacêutica, concentração, população alvo, posologia, indicação terapêutica, entre outros aspectos. O solicitante deve elaborar um relatório clínico que permita o entendimento do conjunto de dados apresentados. O relatório clínico deve conter: Um resumo da metodologia utilizada para busca dos artigos citando bases de dados que foram pesquisadas, critérios de inclusão e exclusão dos artigos e data da pesquisa e; Uma análise criteriosa e consistente do conjunto de estudos apresentados, levando em consideração as variáveis estudadas e sua aplicabilidade e relevância clínica para as indicações terapêuticas propostas. Os resultados dos estudos clínicos apresentados devem ser referentes, especificamente, às concentrações e razões de dose que se pretende registrar. Estudos com resultados favoráveis e desfavoráveis à associação devem estar presentes na análise. Todos os artigos utilizados devem ser apresentados juntamente com o relatório clínico. REFERÊNCIAS BIBLIOGRÁFICAS 1. EUROPEAN MEDICINES AGENCY EMEA. Guideline on Clinical Development of Fixed Combinations Medical Products CHMP/EWP/240/95 Rev. 1. London: Disponível em: Acesso em: 03 mai FOOD AND DRUG ADMINISTRATION FDA. Fixed Dose Combinations, Co-Packaged Drug Products, and Single-EntityVersions of Previously Approved Antiretrovirals for the Treatment of HIV. Maryland: Disponível em: GuidanceComplianceRegulatoryInformation/Guidances/ucm pdf. Acesso em: 03 mai WORLD HEALTH ORGANIZATION WHO. WHO Technical Report Series - Anexo 5 - Guidelines for registration of fixed-dose combination medicinal products - No. 929 Geneva: Disponível em: < Acesso em: 03 mai
219 Posicionamento da Anvisa quanto ao registro de medicamentos novos considerados como me-toos Para esclarecer ao setor regulado, a membros da academia e a seus próprios consultores ad hoc sobre os seus critérios de avaliação para registro de medicamentos novos considerados como me-toos, a Anvisa, por meio da Gerência de Medicamentos Novos, Pesquisa e Ensaios Clínicos (GEPEC), divulga o seguinte posicionamento, o qual se respalda, em parte, em parecer da Câmara Técnica de Medicamentos (CATEME), seu órgão consultivo: Não há consenso sobre o que seja um medicamento me-too, muito embora esta denominação seja de uso comum na literatura internacional, e diversos artigos sobre o tema possam ser encontrados utilizando-se este termo como palavra-chave em pesquisas em bancos de dados como o Medline. Neste contexto, entende-se como me-too um medicamento que embora seja apresentado como inovador não acrescenta nenhum benefício claro, no que diz respeito aos seus perfis de eficácia e segurança, em relação a outros medicamentos já registrados. Por esta definição não são, portanto, considerados como me-toos os medicamentos genéricos ou similares. Existem correntes que defendem que medicamentos classificáveis como me-toos não devam ser registrados, tendo em vista que nada acrescentariam ao que já é disponível no mercado e teriam a desvantagem de não possuir o mesmo acúmulo de evidência de sua segurança. Por esse raciocínio, esses produtos apenas contribuiriam para um inchaço desnecessário do mercado. A Anvisa, porém, diverge desta visão, por considerar difícil, ou mesmo impossível, classificar um medicamento como me-too no momento de seu registro, já que alguns atributos que permitiriam que se fizesse essa qualificação só podem ser verificados depois da comercialização e utilização em larga escala do produto. Entre esses atributos podem ser citados a ocorrência ou não de eventos adversos raros, a identificação de subgrupos de indivíduos que potencialmente se beneficiariam do novo medicamento de forma diferenciada e a descoberta de novas indicações terapêuticas para esse medicamento em particular. Para a Agência, a dificuldade de se classificar um produto como me-too no momento de seu registro impossibilita o estabelecimento de regras que regulem o registro de medicamentos sob tal ótica. Além disso, a legislação brasileira vigente não subsidia o indeferimento de pedidos de registro de medicamentos por este argumento. Por esses motivos, a Anvisa não faz restrição a analisar pedidos de registro de medicamentos que, preliminarmente, pareçam ser metoos e nem respalda o indeferimento de tais pedidos por este motivo. * me-too: eu também, em tradução literal do inglês 219
220 Nota técnica: Reenquadramento de lágrimas artificiais e lubrificantes oculares como medicamentos específicos 1. Conforme determina a Resolução RDC nº 05, de 30 de janeiro de 2015, em seu Art. 1, as lágrimas artificiais e ou lubrificantes oculares são enquadrados na categoria de medicamentos específicos. 2. São consideradas lágrimas artificias e lubrificantes oftálmicos, produtos contendo emolientes, usualmente polímeros solúveis em água (por exemplo, derivados de cellulose, dextrano 70, gelatina, poliois, álcool polivinílico ou povidona), aplicados topicamente no olho para proteger e lubrificar a superfície mucosa, aliviando a irritação, ardor e secura dos olhos. Produtos oftálmicos contendo associações entre emolientes e substâncias farmacologicamente ativas ou substâncias farmacologicamente ativas isoladas permanecem enquadrados em suas categorias atuais. 3. Os requisitos para o registro e a renovação de registro de medicamentos específicos estão dispostos na Resolução RDC n 24, de 14 de junho Desse modo, para o registro dos medicamentos que se enquadrem na definição acima, a empresa deverá protocolar processo endereçada a COGEN/GGMED/SUMED apresentando os seguintes documentos: I - formulários de petição (FP); II - via original do comprovante de recolhimento da taxa de fiscalização de vigilância sanitária, ou isenção, quando for o caso; III - cópia da licença de funcionamento da empresa (alvará sanitário), atualizada, ou protocolo da solicitação da renovação da referida licença; IV - cópia do CRT, atualizado, emitido pelo Conselho Regional de Farmácia; V - cópia do CBPFC, atualizado, emitido pela Anvisa para a linha de produção na qual o medicamento especifico será fabricado; e VI - relatório técnico, contendo as seguintes informações: a relatório de estabilidade do medicamento, conforme legislação vigente; b layout das embalagens primária e secundária, modelo de bula, e rótulo, conforme legislação vigente; c - documentação referente a cada local de fabricação, caso a empresa solicite o registro em mais de um local de fabricação; d - relatório de produção; 220
221 e - controle de qualidade; e f relatório técnico com informações de segurança e eficácia 4. Todos os documentos acima devem ser apresentados conforme determina a RDC 24/2011. Além disso, para que os medicamentos sejam enquadrados como isentos de prescrição médica, deverão também ser observadas as indicações previstas na RDC 138/ As informações sobre segurança e eficácia devem ser comprovadas por meio de relatório de segurança e eficácia não clínica e clínica. No caso de soluções, alternativamente, podem ser apresentados dados de literatura técnico-científica que contemplem essas informações; 6. Os relatórios de eficácia não clínica e clínica devem estar de acordo com a legislação vigente. 7. Em caso de apresentação de dados de literatura para comprovação da eficácia e segurança do medicamento, esses devem contemplar estudos clínicos oriundos de publicação científica indexada, brasileira ou internacional. Os estudos clínicos publicados devem ter sido realizados com formulações cujos princípios ativos sejam os mesmos e tenham sido administrados nas mesmas doses, na mesma posologia, na mesma forma farmacêutica, e pela mesma via de administração pleiteada no registro. Da mesma forma, a indicação de uso do produto deve ser a mesma avaliada no estudo clínico publicado. No caso de associação de princípios ativos, os estudos clínicos publicados devem ter sido realizados com a associação dos princípios ativos nas mesmas doses, na mesma posologia, na mesma forma farmacêutica, mesma via de administração e mesma indicação pleiteada no registro. 8. Para outras formas farmacêuticas que não sejam soluções, deve ser apresentado estudo clínico com o produto objeto do registro. 9. Para os produtos que já estejam registrados como medicamento, os dados de segurança e eficácia devem ser enviados na primeira renovação de registro que ocorrer após a publicação da RDC 05/2015, conforme determina o inciso IX do Art. 48 da RDC 24/ Dadas as características específicas das lágrimas artificiais e lubrificantes oculares, as especificações de densidade, osmolaridade, viscosidade e ph devem ser apresentadas e justificadas tecnicamente no registro ou na renovação de registro do medicamento. Também deve ser informada a massa molar do(s) polímero(s) e devem ser apresentados os resultados do estudo de estabilidade em uso do medicamento. 11. Para os medicamentos que tiveram o registro solicitado antes da publicação da RDC 05/2015 com outro enquadramento que não na classe de específicos, e que ainda não foram avaliados pela Anvisa, serão reenquadrados e avaliados como medicamentos específicos. A documentação faltante será solicitada por meio de notificação de exigência. 221
222 12. Os medicamentos já registrados como novos, similares ou genéricos, passam a ser enquadrados como medicamentos específicos a partir da publicação da RDC 05/2015. Desse modo, a próxima petição a ser realizada junto a Anvisa - alteração pós-registro, renovação, HMP - deverá ser realizada com enquadramento como ESPECÍFICO. As empresas que se enquadrarem nesse caso e tiverem petições de alterações pósregistro em aberto, devem encaminhar, de forma a agilizar a adequação destas petições, via carta endereçada à COGEN/GGMED/SUMED/ANVISA, uma lista contemplando as petições em aberto para que as estas tenham seus assuntos alterados e possam ser avaliadas. 13. Para os medicamentos que tiveram o registro solicitado antes da publicação da RDC 05/2015 com outro enquadramento que não na classe de específicos, e que estão em análise, a análise da petição será transferida para a COGEN e, se necessário, será emitida uma nova exigência para que a empresa apresente os documentos faltantes. O assunto da petição será atualizado e a decisão da solicitação de registro já será publicada como referente ao registro de medicamento específico. Nota técnica: Eficácia e segurança de medicamentos específicos 1. De acordo com o Art. 32 da RDC 24 de 2011, o relatório técnico que acompanha o dossiê de registro de medicamentos específicos deve conter informações sobre segurança e eficácia comprovadas por: I - relatório de segurança e eficácia pré-clínica e clínica; ou II - dados de literatura técnico-científica que contemple essas informações; ou III tradicionalidade de uso. 2. Para a comprovação de segurança e eficácia de acordo com o item I, a empresa deve seguir as normas de pesquisa não clínica e clínica descritas em legislação específica. Os dados devem ser acompanhados de referências bibliográficas quando disponíveis. 2.1.Para a realização dos estudos não clínicos, deve-se seguir o disposto no Guia para a condução de estudos não clínicos de toxicologia e segurança farmacológica necessários ao desenvolvimento de medicamentos. Vale ressaltar que métodos alternativos in vitro validados e aceitos internacionalmente podem ser apresentados em substituição a estudos in vivo. 2.2.Para a realização de ensaios clínicos, deve-se seguir a RDC nº 9/2015, que dispõe sobre o regulamento para a realização de ensaios clínicos com medicamentos no Brasil, e as determinações do Conselho Nacional de Saúde (CNS), estabelecidas por meio da Resolução nº 466/2012 e da Resolução nº 251/ Já para a comprovação de segurança e eficácia de acordo com o item II, são aceitos livros técnico-científicos, informações disponibilizadas em bases científicas eletrônicas, artigos publicados em periódicos indexados e documentos oficiais de instituições governamentais brasileiras e internacionais. Nesses quatro casos, as referências devem ser reconhecidas pela comunidade científica nacional e internacional. Para os livros técnicos científicos, recomenda-se a utilização de handbooks, livros de farmacologia, fitoterapia, fitoquímica, farmacognosia, química medicinal e farmacêutica, formulários farmacêuticos, mementos terapêuticos, e afins. Os artigos científicos devem estar publicados em revistas indexadas pelo sistema WEB QUALIS 222
223 ( As bases científicas eletrônicas aceitas são aquelas indexadas no Portal Periódicos Capes ( A literatura apresentada, no caso de comprovação conforme item II, deve ser referente a ensaios clínicos bem conduzidos, prospectivos, com número significativo de pacientes e preferencialmente, randomizados, controlados e duplos cegos. O(s) estudo(s) deve(m) apresentar resultados positivos estatisticamente significativos para indicação terapêutica proposta. Também são aceitas meta-análises e revisões sistemáticas. O nível de evidência científica das publicações deve ser evidência 1 ou 2 1 e qualidade entre A-B 2 que suportem as indicações propostas. O produto utilizado no ensaio clínico deve conter a mesma entidade química ou insumo farmacêutico ativo (IFA), na mesma forma farmacêutica, posologia e via de administração do produto que se deseja registrar. A apresentação de estudo(s) clínico(s) realizado(s) com o próprio produto é altamente recomendada, principalmente nos casos em que pode haver diferença nas características farmacocinéticas do IFA (por exemplo: polimorfos, IFA micronizado, entre outros) 3.2.Para registro de medicamentos específicos em associações de princípios ativos, devem ser apresentados estudos clínicos referentes à associação nas concentrações, formas farmacêuticas, posologia e vias de administração indicadas no registro com os mesmos requisitos descritos anteriormente ou estudos para os produtos isolados nas concentrações descritas na associação, acompanhados de justificativa técnica que explique a racionalidade da associação ou literatura indexada que demonstre a racionalidade e vantagens da associação dos ativos Não são aceitos estudos de caso, ensaios clínicos com baixa qualidade (C-D), estudos com baixo nível de evidência (3-7), artigos e textos publicados em revistas não indexadas, em livros não científicos ou informações obtidas a partir de sítios eletrônicos diversos Também não são aceitos estudos com partes da molécula ativa ou com moléculas assemelhadas à molécula ativa ou com moléculas da mesma classe da molécula ativa, mas que não são referentes à molécula ativa presente no medicamento. Por exemplo, para comprovação de eficácia de um medicamento que contenha aspartato de arginina, não são aceitos estudos realizados com glutamina, com aminoácidos em geral, com cloridrato de arginina, ou com aspartilarginina Além dos estudos clínicos de eficácia e segurança, recomenda-se que seja apresentada comprovação de registro do(s) mesmo(s) IFA(s), na mesma forma farmacêutica, na mesma indicação e concentração em outro país; dados sobre os aspectos farmacocinéticos especiais do princípio ativo (uso pediátrico, geriátrico, na gravidez e puerpério, entre outros); dados sobre as interações medicamentosas efetivamente estudadas e potenciais; dados sobre os aspectos farmacodinâmicos e farmacocinéticos (absorção, distribuição, metabolismo e excreção) do princípio ativo, e dados sobre segurança e índice terapêutico do fármaco, quando conhecido. 4. Para fins de comprovação de segurança e eficácia por tradicionalidade de uso, deverá ser observado o disposto no Art. 39 da RDC nº 24 de 14 de junho de Devendo ser apresentadas publicações técnicocientíficas que serão avaliadas conforme os seguintes critérios: I - indicação de uso episódico ou para curtos períodos de tempo; 223
224 II - indicação para doenças de baixa gravidade ou relacionada à melhoria ou manutenção da saúde; III - coerência das indicações terapêuticas propostas com as comprovadas pelo uso tradicional; IV - ausência de grupos ou substâncias químicas tóxicas, ou presentes dentro de limites comprovadamente seguros; V - comprovação de continuidade de uso seguro por período igual ou superior a 10 anos no Brasil; e VI - racionalidade das associações de ativos. 1 De acordo com Melnyk et al., 2005, a qualidade das evidências é classificada em sete níveis. No nível 1, as evidências são provenientes de revisão sistemática ou meta-análise de ensaios clínicos relevantes randomizados controlados ou oriundas de diretrizes clínicas baseadas em revisões sistemáticas de ensaios clínicos randomizados controlados; nível 2, evidências derivadas de pelo menos um ensaio clínico randomizado controlado bem delineado; nível 3, evidências obtidas de ensaios clínicos bem delineados sem randomização; nível 4, evidências provenientes de estudos de coorte e de caso-controle bem delineados; nível 5, evidências originárias de revisão sistemática de estudos descritivos e qualitativos; nível 6, evidências derivadas de um único estudo descritivo ou qualitativo; nível 7, evidências oriundas de opinião de autoridades e/ou relatório de comitês de especialistas. 2 De acordo com o National Health Service da Grã-Bretanha (adaptado): A - Estudos experimentais ou observacionais de melhor consistência. B - Estudos experimentais ou observacionais de menor consistência. C - Relatos de casos estudos não controlados. D - Opinião desprovida de avaliação crítica, baseada em consensos, estudos fisiológicos ou modelos animais. 224

225 225

226 226
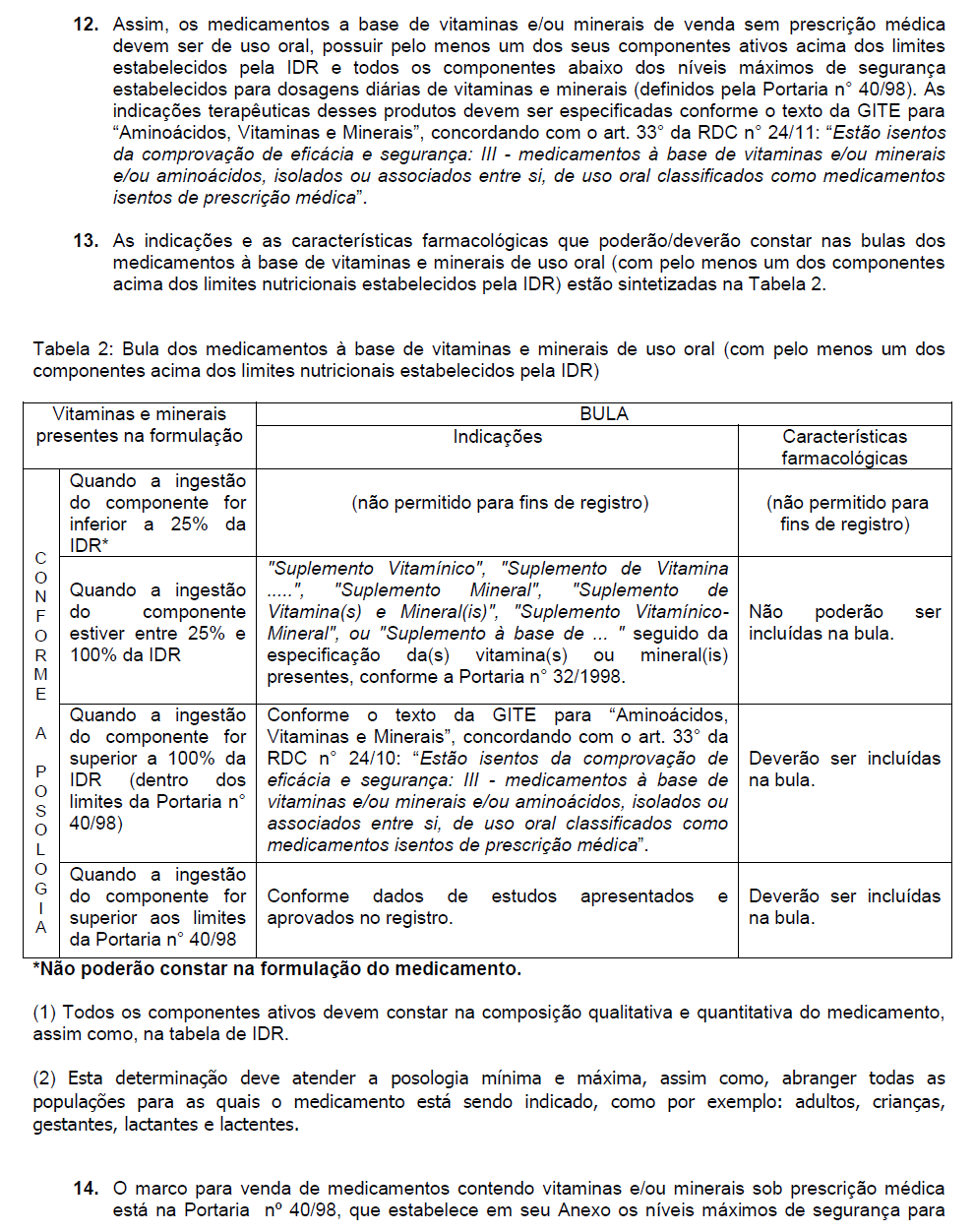
227 227

228 228
229 Orientação de serviço 03/2014 GGMED/SUMED/ANVISA Esclarecimentos e prorrogação de prazo relacionados à Orientação deserviço 01/2014, que trata do registro e pós-registro de medicamentos específicos: suplementos vitamínicos e minerais de uso oral 1. Com o objetivo de esclarecer dúvidas e questionamentos que surgiram em razão da publicação da Orientação de Serviço nº 01/2014 GGMED/SUMED/ANVISA, que trata do registro e pós-registro de medicamentos específicos: suplementos vitamínicos e minerais de uso oral, informamos: a. Os medicamentos contendo vitaminas e minerais com indicação de ingestão entre 25% a 100% da IDR poderão mencionar, nas bulas, dentro da seção COMO ESTE MEDICAMENTO FUNCIONA? as funções de "Suplemento Vitamínico", "Suplemento de Vitamina..."Suplemento Mineral", "Suplemento de Vitamina(s) e Mineral(is)", "Suplemento Vitamínico-Mineral", ou "Suplemento à base de...".é permitida a citação de funções cientificamente comprovadas das vitaminas e minerais com a descrição do papel fisiológico desses nutrientes no desenvolvimento ou manutenção do organismo, conforme previsto na Portaria nº 32/1998. b. A rotulagem e a bula do medicamento devem ser adequadas de modo a excluir do texto de bula e rotulagem quaisquer referências às indicações terapêuticas e características farmacológicas de vitaminas e minerais com indicação de ingestão inferior a 25% da IDR. No caso de informações sobre advertência e contraindicação de vitaminas e minerais com indicação de ingestão inferior a 25% da IDR, estas deverão ser mantidas no texto de bula e rotulagem. c. As bulas dos produtos deverão informar as vitaminas e/ou minerais presentes em quantidade inferior a 25% da IDR sem citar a quantidade, considerando a posologia recomendada, listando-as, abaixo das substâncias ativas, como "outros componentes", na seção reservada às informações relativas à composição do medicamento. As embalagens dos referidos produtos poderão, a critério da empresa, informar a presença de tais substâncias, da mesma forma descrita para as bulas. d. Os produtos que mencionem a Denominação Comum Brasileira (DCB) do ativo em quantidade inferior a 25% da IDR abaixo do nome comercial do produto devem retirar essa informação abaixo do nome comercial no texto de bula e rotulagem. e. Nos casos em que vitaminas e minerais abaixo dos 25% sejam quantificados como marcadores em testes de dissolução, a empresa deverá substituir a vitamina e/ou mineral eleito como marcador por outro adequado que esteja em concentração superior a 25% da IDR, conforme recomendações vigentes em compêndios reconhecidos pela Anvisa. Caso a empresa possua estudos de estabilidade de medicamentos já registrados em andamento, sugerimos que seja quantificado novo marcador, mas que o acompanhamento do marcador anterior permaneça até o final do estudo. f. Os medicamentos de venda sob prescrição que possuam estudo clínico com o seu produto ou publicações de estudos clínicos com produtos de mesma composição quali-quantitativa, mesma forma farmacêutica e mesma via de administração, que comprovem a atividade terapêutica descrita em bula, não precisarão modificar a indicação e posologia, desde que essas sejam suportadas pelos estudos/publicações. A empresa deverá, nesse caso, peticionar os assuntos previstos na OS 01/2014 GGMED/SUMED/ANVISA, com justificativa para a não realização das alterações de texto de bula e de rotulagem, acompanhada dos estudos disponíveis. 229
230 g. As empresas detentoras de registro de produtos com nomes comerciais que façam menção a ativos que estejam presentes na formulação em quantidade inferior a 25% da IDR devem contatar a Coordenação de Medicamentos Específicos, Notificados e Gases Medicinais (COGEN) para avaliação individualizada anteriormente a adequação de bula e rotulagem do produto. h. As petições de renovação de registro e de pós-registro protocoladas anteriormente à vigência da norma de adequação de formulação a ser publicada, serão avaliadas pela Anvisa seguindo o disposto no texto da OS 01/2014 GGMED/SUMED/ANVISA. Desse modo, para os medicamentos já registrados, não será solicitada a adequação de formulação antes da publicação de normativa a respeito. i. A norma de adequação de formulação a ser publicada estabelecerá um prazo para adequação por parte das empresas. Portanto nesse primeiro momento, a empresa deverá proceder apenas com a adequação de texto de bula e rotulagem. j. Caso a empresa assim o deseje, poderá avaliar o seu produto e optar por alterar a posologia ou a população-alvo, desde que justificadas, e peticionar tal alteração em substituição às alterações de texto de bula e rotulagem previstas pela OS 01/2014 GGMED/SUMED/ANVISA, dentro do prazo máximo estabelecido neste documento. Nesse caso, não será necessária a adequação de formulação futura. 2. Dessa forma, considerando os esclarecimentos prestados, as solicitações recebidas pela Anvisa e o texto da OS 1/2014 GGMED/SUMED/ANVISA, as empresas deverão peticionar os assuntos: 10606: ESPECÍFICO - Notificação de alteração de texto de bula adequação dos medicamentos com princípios ativos (vitaminas, minerais e/ou aminoácidos) abaixo de 25% da IDR e também 10607: ESPECÍFICO Notificação de alteração de rotulagem adequação dos medicamentos com princípios ativos (vitaminas, minerais e/ou aminoácidos) abaixo de 25% da IDR. a. As notificações de alteração de texto de bula e rotulagem deverão ser protocoladas até 30 de novembro de Após o protocolo, deverá ser considerado o prazo de 180 dias da RDC nº 47/09 e da RDC nº 71/09 para esgotamento de estoque. b. As empresas que já protocolaram as alterações de texto de bula e de rotulagem, terão até o dia 30 de novembro de 2014 para protocolarem, sob a forma de aditamento, documentação complementar que julgarem necessária. Gerência Geral de Medicamentos GGMED/SUMED/ANVISA 230
231 Nota Técnica nº 17/2015/COGEN/GGMED/SUMED/ANVISA Assunto Mudanças relacionadas ao Insumo Farmacêutico Ativo para Medicamentos Específicos 1. Conforme determina o parágrafo único do Art. 120 da RDC 48/2009, as petições referentes a Mudanças relacionadas ao Fármaco, descritas no Capítulo XII da RDC 48/2009, não se aplicam à categoria de medicamentos específicos. 2. Entretanto, a RDC 17/2010, que dispõe sobre as Boas Práticas de Fabricação de Medicamentos, estabelece, no Art. 305, que: A estabilidade de um produto deve ser determinada antes da comercialização e deve ser repetida após quaisquer mudanças significativas nos processos de produção, equipamentos, materiais de embalagem e outras que possam influir na estabilidade do produto. 3. Adicionalmente, no Guia para a Realização de Estudos de Estabilidade, anexo da RE n 01/2005, temos que: A estabilidade de produtos farmacêuticos depende de fatores ambientais como temperatura, umidade e luz, e de outros relacionados ao próprio produto como propriedades físicas e químicas de substâncias ativas e excipientes farmacêuticos, forma farmacêutica e sua composição, processo de fabricação, tipo e propriedades dos materiais de embalagem. 4. Ainda conforme disposto no Art. 159 da RDC 17/2010, matérias-primas a serem utilizadas para produção de medicamentos devem ser adquiridas somente de fornecedores aprovados pela empresa. No Art. 215 da RDC 17/2010, também fica estabelecido que a frequência de execução de ensaios em cada matéria-prima deve ser determinada de acordo com a estabilidade da matéria-prima. As disposições sobre realização estudos de estabilidade de Insumos Farmacêuticos Ativos (IFA) estão estabelecidas na RDC 45/ Considerando que essas normas aplicam-se a todas as categorias de medicamentos, inclusive os medicamentos específicos, fica evidente que a alteração do fabricante de IFA do medicamento específico, além de requerer nova qualificação de fornecedores, e, possivelmente o estabelecimento de novas especificações para a matéria-prima, pode impactar na qualidade do medicamento, de modo que a empresa produtora do medicamento deve demonstrar que o produto fabricado a partir de determinado fabricante de IFA não sofrerá alterações que possam prejudicar sua qualidade, e afetar a eficácia e segurança deste medicamento. 6. Quanto às alterações pós-registro relacionadas ao IFA, os medicamentos específicos se dividem em dois grupos: a. Para medicamentos com substâncias ativas que se enquadram no inciso VII do artigo 5º da RDC 24/2011 (medicamentos a base de rutina, quercetina, hesperidina, diosmina, troxerrutina e cumarina, isolados ou associados entre si), conforme o Art. 47 da RDC 24/2011, se aplica o capítulo XII da RDC 48/2009 e, portanto, caso queria realizar alguma alteração ou inclusão referente ao IFA, a 231
232 empresa deverá protocolar petição com os assuntos ESPECÍFICO - Alteração da rota de síntese do fármaco, ESPECÍFICO - Alteração de local de fabricação do fármaco, ESPECÍFICO - Inclusão de rota de síntese de fármaco ou ESPECÍFICO - Inclusão de local de fabricação do fármaco. Neste caso as alterações somente poderão ser implementadas após o deferimento da petição pela Anvisa publicado no Diário Oficial da União (DOU). b. Para os demais medicamentos específicos, conforme já informado no documento Orientações para alterações pós-registro em medicamentos sintéticos, disponível no portal da Anvisa ( não se aplica o protocolo de inclusão de local de fabricação de fármaco conforme RDC 48/2009. Este documento também estabelece que A empresa deverá informar no HMP do produto, como informação suplementar, o novo local de fabricação do fármaco e fornecer as informações técnicas sobre o mesmo. Conforme determina o Art. 210 da RDC 48/2009: Nos casos não previstos nesta Resolução, ou que não satisfaçam a algum dos quesitos especificados, ficará a critério da Anvisa estabelecer os testes e a documentação que deverão ser apresentados.. Desse modo, tendo em vista que, conforme disposto acima, a alteração de local de fabricação ou de rota de síntese ou extração do IFA pode impactar na qualidade, na segurança e na eficácia do medicamento, e, de modo facilitar o entendimento sobre a documentação necessária em casos de alteração relacionadas ao IFA não previstos no Art. 47 da RDC 24/2011, informamos que a empresa deverá proceder da seguinte forma: i. Nos casos em que a alteração referente ao IFA não implicar em nenhuma outra alteração de pós-registro passível de peticionamento de acordo com a RDC 48/2009, a empresa deverá informar a alteração via Histórico de Mudanças do Produto (HMP), conforme já previsto no documento Orientações para alterações pós-registro em medicamentos sintéticos, incluindo a seguinte documentação: 1. Perfil comparativo de impurezas entre 1(um) lote do IFA obtido pela rota de síntese, ou extração ou local de fabricação aprovada no registro e 1(um) lote do IFA obtido pela nova rota de síntese, extração ou local de fabricação ou justificativa da ausência; 2. Rota de síntese ou de extração completa com produtos intermediários ou justificativa da ausência; 3. Laudo analítico de controle de qualidade do IFA referente a 1(um) lote emitido pelo fabricante do medicamento; 4. Laudo analítico de controle de qualidade do IFA referente a 1(um) lote emitido pelo fabricante do IFA; 5. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; 6. Protocolo de estudo de estabilidade de 1(um) lote do produto acabado realizado com o novo IFA; 7. Relatório de estudo de estabilidade de 1(um) lote do produto acabado realizado com o novo IFA (a ser incluído nos HMP dos anos seguintes à implementação da alteração); 232
233 8. Informações referentes aos solventes utilizados; à lista de solventes residuais; ao polimorfismo; aos limites, quantificação e especificação de impurezas de síntese e a produtos de degradação, além das informações referentes à quiralidade e proporção de isômeros, conforme o caso; 9. Discussão relativa ao impacto de eventuais alterações do perfil de impureza e validação, ou revalidação, da metodologia de análise. ii. Nos casos em que a alteração referente ao IFA implicar em alguma outra alteração pósregistro passível de peticionamento de acordo com a RDC 48/2009, a empresa deverá peticionar a alteração pós-registro correspondente e informar a alteração referente ao IFA com alteração concomitante, incluindo a documentação citada no inciso i. 7. Qualquer alteração de especificação da matéria-prima decorrente da alteração referente ao IFA deve ser peticionada como Alteração de especificação e método analítico. Caso haja mais de uma alteração decorrente da alteração referente ao IFA, tais como alterações de processo, alterações de excipientes, dentre outras, estas devem ser peticionadas como alterações concomitantes ou paralelas, nos termos da RDC 48/ Os estudos de estabilidade realizados com o IFA devem seguir o disposto na RDC 45/2012 e podem ser realizados pelo fabricante do IFA ou pelo fabricante do medicamento, devendo estar disponíveis para avaliação pelas autoridades sanitárias em momento oportuno. 9. Qualquer resultado fora da especificação observado durante a realização dos estudos de estabilidade deve ser informado imediatamente à Anvisa e a empresa deve tomar as ações necessárias caso haja evidências de que a estabilidade do medicamento tenha sido reduzida pela alteração referente ao IFA, conforme determina o Art. 12 da RDC 48/2009. Gerência Geral de Medicamentos GGMED/SUMED/ANVISA 233
234 RESOLUÇÃO RDC Nº 199, DE 26 DE OUTUBRO DE 2006 (*) A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 354 da ANVISA, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada em 23 de outubro de 2006, e considerando o disposto no Art. 41 2º da Lei nº 9782 de 1999, alterada pela Medida Provisória de 2001; considerando a definição de medicamento presente no Art. 4º inciso II da Lei 5991 de 1973; considerando o art. 2 inciso III da Lei nº 9279/96, que regula os direitos e obrigações relativos à propriedade industrial; adota a seguinte Resolução de Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Para efeito desta Resolução consideram-se as seguintes definições: MEDICAMENTO DE NOTIFICAÇÃO SIMPLIFICADA - produto farmacêutico, tecnicamente obtido ou elaborado, com finalidade profilática, curativa ou paliativa na qual existe baixo risco de que seu uso ou exposição possa causar conseqüências e ou agravos à saúde quando observadas todas as características de uso e de qualidade descritas no Anexo I desta Resolução. NOTIFICAÇÃO - comunicação à autoridade sanitária federal (ANVISA) referente à fabricação, importação e comercialização dos medicamentos de notificação simplificada relacionados no Anexo I deste regulamento. AFE - AUTORIZAÇÃO DE FUNCIONAMENTO DE EMPRESA- Ato privativo do órgão ou da entidade competente do Ministério da Saúde, incumbido da vigilância sanitária dos produtos de que trata este Regulamento, contendo permissão para que as empresas exerçam as atividades sob regime de vigilância sanitária, instituído pela Lei nº 6.360, de 1976, mediante comprovação de requisitos técnicos e administrativos específicos. Art. 2º Fica instituída a notificação simplificada de medicamentos por meio eletrônico disponível no site da ANVISA. 1º Para efeito deste regulamento são considerados medicamentos de notificação simplificada aqueles constantes no Anexo I. Art 3º A notificação não exime as empresas das obrigações do cumprimento das Boas Práticas de Fabricação e Controle e das demais regulamentações sanitárias. 234
235 1º os medicamentos sujeitos à notificação simplificada devem adotar, integralmente, as informações padronizadas no Anexo I deste regulamento. 2º Os produtos no Anexo I são de venda isenta de prescrição médica. 3º É vedada a comercialização dos produtos do Anexo I na forma farmacêutica injetável. 4º Todos os produtos que contém cânfora como principio ativo são passíveis de registro devendo seguir os mesmos critérios adotados para registro de Medicamentos Específicos. Excetuam-se os casos de associações com princípios ativos enquadrados em outras categorias existentes. 5º As inclusões, alterações e exclusões do Anexo I serão publicadas periodicamente pela ANVISA, em resolução específica, após avaliação das informações apresentadas pelas empresas através do requerimento presente no anexo III deste regulamento. A ANVISA poderá solicitar bibliografia complementar, a seu critério, para auxiliar na decisão de inclusão, alteração ou exclusão da formulação solicitada. Art. 4º Apenas as empresas fabricantes, que cumprem as Boas Práticas de Fabricação e Controle, de acordo com a legislação vigente, e que estão devidamente autorizadas/licenciadas pela Autoridade Sanitária competente, podem notificar e fabricar os produtos abrangidos por esta Resolução,mediante o Certificado de Boas Praticas de Fabricação e Controle( CBPFC) ou protocolo de solicitação do pedido de CBPFC com status satisfatório no Banco de dados de Inspeção da ANVISA. Art. 5º Os estudos de estabilidade devem ser realizados conforme GUIA DE ESTABILIDADE. Quando houver inviabilidade técnica para realização dos testes requeridos a empresa deverá apresentar justificativa arrazoando os motivos técnicos. Art. 6º A notificação dos produtos listados no Anexo I deve ser precedida pela notificação da produção de lotes piloto de acordo com o GUIA PARA NOTIFICAÇÃO DE LOTE PILOTO, exceto para produtos que possuem cadastro ou registro vigente junto à Anvisa. 1º Os lotes piloto poderão ser comercializados, a critério do fabricante, após a realização do estudo de estabilidade acelerado e a devida notificação do produto, conforme estabelecido neste regulamento. Art. 7º A notificação deve seguir os seguintes critérios: 1º A notificação deve ser realizada, exclusivamente, pela empresa com autorização de funcionamento para fabricar e/ou importar medicamentos. 2º A empresa deverá realizar uma notificação individual para cada produto, conforme este regulamento. 3º A empresa deverá atualizar a notificação sempre que houver modificação em quaisquer informações prestadas por meio da notificação eletrônica. 4º Todas as notificações devem ser renovadas a cada 5 (cinco) anos, mediante nova notificação de cada produto, respeitando os prazos estabelecidos no Art. 12 da Lei nº 6.360/
236 5º Quando houver suspensão da fabricação do produto, a empresa deverá notificar a exclusão de comercialização deste produto, mediante peticionamento eletrônico. 6º As notificações de que trata o caput deste artigo estão isentas do pagamento de taxa. 7º Será disponibilizada, para consulta no site da ANVISA, a relação de empresas e produtos notificados, imediatamente após a realização da notificação. Art. 8º Os medicamentos de notificação simplificada devem possuir, em sua rotulagem, o enquadramento nesta Resolução, adotando a frase: " MEDICAMENTO DE NOTIFICAÇÃO SIMPLIFICADA RDC nº /2006. AFE nº:...". 1º A rotulagem dos produtos objeto deste regulamento deve seguir o estabelecido no Anexo I e no Anexo II, ficando dispensados de apresentação de bula. 2º Fica facultada a utilização de embalagem secundária, caso constem na embalagem primária todas as informações exigidas no Anexo I e Anexo II deste regulamento. As informações sobre especificações analíticas mínimas e referência não devem constar na rotulagem do produto. 3º Fica dispensada a utilização de tinta reativa na rotulagem de produtos desta categoria, porém as embalagens devem apresentar lacre ou selo de segurança, para garantia da inviolabilidade do produto. 4º Estes produtos devem adotar para sua identificação, o nome do produto ou sinônimo presentes no Anexo I deste regulamento, sendo facultada a adoção de marca ou nome comercial. Art. 9º A adequação a este regulamento de medicamentos cadastrados ou registrados na ANVISA deve ser realizada respeitando as seguintes disposições: I -Todos os produtos cadastrados na ANVISA como isentos de registro devem se adequar a este regulamento no momento de sua renovação. A critério da empresa, a adequação a esta Resolução poderá ser realizada antes do período de renovação. II - Os produtos listados no Anexo I, porém atualmente registrados em outras categorias de medicamentos, devem se adequar a este regulamento no momento de sua renovação. A critério da empresa, a adequação a esta Resolução poderá ser realizada antes do período de renovação. 1º As petições referentes a cadastro de medicamentos isentos de registro em análise ou em arquivamento temporário serão encerradas a partir da vigência deste regulamento. No caso de petições de renovação de cadastro de medicamentos, protocoladas antes da publicação deste regulamento, a adequação deve ocorrer em até 180 dias. 2º Caso haja produtos registrados ou cadastrados com indicações diferentes, a empresa deverá adequar-se as informações existentes no Anexo I e posteriormente, providenciar protocolo do requerimento de inclusão, alteração ou exclusão presente no Anexo III deste regulamento e aguardar a publicação. 3º O cadastro de medicamentos, cujo princípio ativo, concentração e/ou forma farmacêutica não estão relacionados no Anexo I deste regulamento, são válidos até o término de sua vigência, devendo 236
237 posteriormente enquadrar-se a essa Resolução ou aos regulamentos para registro de medicamentos junto a Anvisa. Art. 10 As informações apresentadas na Notificação são de responsabilidade da empresa e serão objeto de controle sanitário pela ANVISA. Art. 11 Ficam revogados art. 3º e art. 8º da Resolução RDC nº 132, de 29 de maio de 2003, e os itens ; 3.7 e 7.1 do anexo da Resolução RDC nº 333, de 19 de novembro de Art. 12 Esta Resolução entrará em vigor 15 dias da publicação ANEXO I - LISTA PADRONIZADA DE MEDICAMENTOS SUJEITOS A NOTIFICAÇÃO SIMPLIFICADA I - Serão objetos de notificação os medicamentos apresentados neste Anexo. II - As especificações analíticas para o produto acabado devem estar de acordo com monografia inscrita em compêndio oficialmente reconhecido pela Anvisa. III - Na ausência de monografia oficial para o produto acabado, deverão ser realizados os testes descritos nos métodos gerais da Farmacopéia Brasileira, e demais testes necessários, desenvolvidos pelo fabricante, para garantir a qualidade do medicamento. Todo laudo de análise de controle de qualidade do produto acabado, independente da forma farmacêutica, deve apresentar, no mínimo, as seguintes informações ou justificativa técnica de ausência: Características organolépticas/aparência; Identificação e teor do(s) princípio(s) ativo(s); Limites microbianos: contagem de bactérias e fungos totais e pesquisa de patógenos. a) Para as formas farmacêuticas sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: Desintegração; Dissolução; Dureza; Peso médio; Umidade. b) Para as formas farmacêuticas líquidas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: ph; Densidade; Viscosidade; Volume ou peso médio. 237
238 238
239 239
240 240
241 241
242 242
243 243
244 244
245 245
246 246
247 247
248 248
249 249
250 250
251 251
252 252
253 253
254 254
255 255
256 256
257 257
258 258
259 259
260 260
261 261
262 262
263 263
264 264
265 265
266 266
267 ANEXO II - MODELO DE ROTULAGEM DE MEDICAMENTOS DE NOTIFICAÇÃO SIMPLIFICADA Nome comercial (FACULTATIVO) Nome do produto ou sinônimo (conforme Anexo I) Concentração do princípio ativo (conforme Anexo I) Forma farmacêutica (conforme Anexo I) Via de administração Uso (adulto, pediátrico, adulto e pediátrico) Conteúdo da embalagem Composição: Nome do princípio ativo...concentração Excipientes (relacionar sem mencionar concentração na fórmula) É facultado a inclusão de informações adicionais voltadas para características organolépticas. Indicação (conforme Anexo I) Modo de Usar (conforme Anexo I) Advertência (conforme Anexo I) Advertências específicas do produto conforme legislação vigente Cuidados de Conservação Frase "TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DE CRIANÇAS" Frase "Para correta utilização deste medicamento, solicite orientação do farmacêutico." Frase "MEDICAMENTO DE NOTIFICAÇÃO SIMPLIFICADA RDC Nº... de AFE nº :...". Frase "AO PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO" Nome do Farmacêutico Responsável e respectivo número de CRF Nome da empresa notificadora Número de CNPJ da empresa notificadora Endereço completo da empresa notificadora 267
268 Fabricado por: (quando for o caso) Nome da empresa fabricante Número de CNPJ da empresa fabricante Endereço completo da empresa fabricante Número do SAC da empresa notificadora Número de Lote Data de Fabricação Prazo de Validade Código de barras ANEXO III - REQUERIMENTO PARA INCLUSÃO, ALTERAÇÃO OU EXCLUSÃO DE MEDICAMENTOS OU INFORMAÇÕES PRESENTES NO ANEXO I Dados do solicitante: Nome do solicitante (jurídica ou física): Endereço: FAX: Telefone: Dados do produto: Princípio Ativo: Concentração: Forma farmacêutica: ( )INCLUSÃO ( ) produto Preencher todos os campos: Sinônimo Indicação Modo de Usar Advertência Referência bibliográfica Referência bibliográfica Referência bibliográfica Referência bibliográfica 268
269 Especificações analíticas mínimas Referência bibliográfica ( ) informações sobre produto já existente no Anexo I Preencher somente o campo pertinente: Sinônimo Indicação Modo de Usar Advertência Especificações analíticas mínimas Referência bibliográfica Referência bibliográfica Referência bibliográfica Referência bibliográfica Referência bibliográfica ( ) EXCLUSÃO ( ) produto ( ) infomações sobre produto já existente no Anexo I ( )sinônimo ( )Indicação ( )modo de usar ( )advertência ( )especificações analíticas mínimas Justificativa Referência Bibliográfica ( ) ALTERAÇÃO ( ) nome do produto ( )princípio ativo ( )concentração ( )forma farmacêutica ( )sinônimo ( )Indicação ( )modo de usar ( )advertência ( )especificações analíticas mínimas Justificativa 269
270 Referência Bibliográfica (*) Republicada por ter saído no DOU nº 208, de , Seção 1, pág. 167, com incorreção no original. 270
271 INSTRUÇÃO NORMATIVA Nº 03, DE 28 DE ABRIL DE 2009 Dispõe sobre a atualização do Anexo I da Resolução - RDC No- 199, de 26 de outubro de 2006, e dá outras providências. O Diretor-Presidente da Agência Nacional de Vigilância Sanitária, no uso das atribuições que lhe conferem o Decreto de nomeação, de 4 de janeiro de 2008, do Presidente da República, e o inciso X do art. 13 do Regulamento da ANVISA, aprovado pelo Decreto n 3.029, de 16 de abril de 1999, tendo em vista o disposto no inciso VIII do art. 16 e no inciso II, 2º do art. 55 do Regimento Interno da ANVISA, aprovado nos termos do Anexo I da Portaria No- 354, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, e, considerando o disposto no art 3º, 5, da Resolução RDC n.º 199, de 26 de outubro de 2006; considerando a necessidade de atualizar o Anexo I da Resolução RDC n.º 199, de 2006, considerando que a matéria foi submetida à apreciação da Diretoria Colegiada que a aprovou em reunião realizada em 22 de abril de 2009, resolve: Art. 1º Determinar a publicação da atualização do Anexo I da Resolução RDC n.º 199, de 26 de outubro de Art. 2 Esta Instrução Normativa entra em vigor na data de sua publicação. DIRCEU RAPOSO DE MELLO ANEXO I LISTA PADRONIZADA DE MEDICAMENTOS SUJEITOS A NOTIFICAÇÃO SIMPLIFICADA I - Serão objetos de notificação os medicamentos apresentados neste Anexo. II - As especificações analíticas para o produto acabado devem estar de acordo com monografia inscrita em compêndio oficialmente reconhecido pela Anvisa. III - Na ausência de monografia oficial para o produto acabado, deverão ser realizados os testes descritos nos métodos gerais da Farmacopéia Brasileira, e demais testes necessários, desenvolvidos pelo fabricante, para garantir a qualidade do medicamento. Todo laudo de análise de controle de qualidade do produto acabado, independente da forma farmacêutica, deve apresentar, no mínimo, as seguintes informações ou justificativa técnica de ausência: Características organolépticas/aparência; Identificação e teor do(s) princípio(s) ativo(s); Limites microbianos: contagem de bactérias e fungos totais e pesquisa de patógenos. a) Para as formas farmacêuticas sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: Desintegração; Dissolução; Dureza; Peso médio; Umidade. b) Para as formas farmacêuticas líquidas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: ph; Densidade; 271
272 Viscosidade; Volume ou peso médio. 272
273 273
274 274

275 275
276 276
277 RDC Nº 47, DE 08 DE SETEMBRO DE 2009 (NÃO APLICÁVEL A MEDICAMENTOS NOTIFICADOS) Estabelece regras para elaboração, harmonização, atualização, publicação e disponibilização de bulas de medicamentos para pacientes e para profissionais de saúde. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária - Anvisa, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 354 da Anvisa, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada 04 em agosto de 2009; considerando que a saúde é direito de todos e dever do Estado, garantido mediante políticas sociais e econômicas que visem à redução do risco de doença e de outros agravos e ao acesso universal e igualitário às ações e serviços para sua promoção, proteção e recuperação, nos termos do art. 196 da Constituição da República Federativa do Brasil, de 5 de outubro de 1988; considerando a Lei nº 6.360, de 23 de setembro de 1976, e o Decreto nº , de 5 de janeiro de 1977, que dispõe sobre o sistema de vigilância sanitária a que ficam sujeitos os medicamentos; considerando a Lei nº 9.787, de 10 de fevereiro de 1999, que altera a Lei nº 6.360, de 23 de setembro de 1976, que dispõe sobre o medicamento genérico e sobre a utilização de nomes genéricos em produtos farmacêuticos; considerando a Lei nº 5.991, de 17 de dezembro de 1973, e o Decreto nº , de 10 de junho de 1974 que dispõe sobre o controle sanitário do comércio de drogas, medicamentos, insumos farmacêuticos e correlatos; considerando a Lei nº 6.437, de 20 de agosto de 1977, que dispõe sobre as infrações à legislação sanitária federal e estabelece as respectivas penalidades; considerando o direito à informação, às pessoas assistidas, sobre sua saúde conforme previsto nos termos do inciso V do art. 7º da Lei Orgânica da Saúde (LOS), Lei nº 8.080, de 19 de setembro de 1990; considerando o direito à informação adequada e clara sobre os diferentes produtos e serviços, com especificação correta de quantidade, características, composição, qualidade e preço, bem como sobre os riscos que apresentem, conforme o previsto pelo inciso III do art. 6 do Código de Defesa do Consumidor, Lei nº 8078, de 11 de setembro de 1990; considerando que compete à União cuidar da saúde e assistência pública, da proteção e garantia das pessoas portadoras de deficiência, nos termos do inciso II do art. 23 da Constituição; considerando as disposições previstas pela Lei n , de 8 de novembro de 2000, que dá prioridade de atendimento às pessoas que especifica, e dá outras providências; considerando as disposições previstas pela Lei n , de 19 de dezembro de 2000, que estabelece normas gerais e critérios básicos para a promoção da acessibilidade das pessoas portadoras de deficiência ou com mobilidade reduzida, e dá outras providências; considerando as disposições previstas pelo Decreto nº 5.296, de 02 de dezembro de 2004, que Regulamenta a Lei n , de 8 de novembro de 2000, que dá prioridade de atendimento às pessoas que especifica, e a Lei n , de 19 de dezembro de 2000, que estabelece normas gerais e critérios básicos para a promoção da acessibilidade das pessoas portadoras de deficiência ou com mobilidade reduzida, e dá outras providências; considerando as diretrizes estabelecidas pela Comissão Brasileira de Braille - CBB, e pela Associação Brasileira de Normas Técnicas - ABNT, quanto a utilização do sistema Braille; 277
278 considerando a Lei n 8.926, de 9 de agosto de 1994, que torna obrigatória a inclusão, nas bulas de medicamentos, de advertências e recomendações sobre seu uso por pessoas de mais de 65 anos; considerando o documento Standard Rules on the Equalization of Opportunities for Persons with Disabilities adotado pela Assembléia Geral das Organizações das Nações Unidas; considerando as diretrizes, as prioridades e as responsabilidades estabelecidas na Política Nacional de Medicamentos, instituída pela Portaria n 3.916/MS/GM, de 30 de outubro de 1998, e aprovada pelo CNS pela Resolução n 338, de 20 maio de 2004, que busca garantir condições para segurança e qualidade dos medicamentos utilizados no país, promover o uso racional e o acesso da população àqueles considerados essenciais; considerando a importância do acesso à informação imparcial e de qualidade para orientar o autocuidado e a automedicação disposta no Report of the 4th WHO - Consultative Group on the Role of the Pharmacist; considerando que as informações sobre medicamentos devem orientar pacientes e profissionais de saúde, favorecendo o uso racional de medicamentos, as bulas devem ser elaboradas com alto padrão de qualidade, com informações imparciais e fundamentadas cientificamente, mesmo quando estiverem dispostas em linguagem simplificada; considerando que as bulas de medicamentos no mercado devem ser reavaliados e harmonizados em face da heterogeneidade e assimetria de informações; considerando a necessidade de harmonizar a forma e o conteúdo das bulas de todos os medicamentos registrados e comercializados no Brasil e unificar a regulamentação sobre o assunto; considerando a competência da Anvisa, no cumprimento de suas atribuições regulamentares, quanto a implementação de ações para agilizar a operacionalização de suas atividades administrativas quanto ao registro, atualização e revalidação de produtos; considerando a Medida Provisória nº , de 23 de agosto de 2001 que instituiu a isenção do recolhimento de taxa para acréscimo ou alteração de registro, referente a texto de bula; adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Fica aprovado o Regulamento Técnico que estabelece os requisitos mínimos para elaboração, harmonização, atualização, publicação e disponibilização de bulas de medicamentos para pacientes e para profissionais de saúde. CAPÍTULO I - DAS DISPOSIÇÕES INICIAIS Seção I - Objetivo Art. 2º Este Regulamento possui o objetivo de aprimorar a forma e o conteúdo das bulas de todos os medicamentos registrados e notificados, comercializados no Brasil, visando garantir o acesso à informação segura e adequada em prol do uso racional de medicamentos. Seção II - Abrangência Art. 3º Este Regulamento se aplica a todos os medicamentos registrados ou notificados na Anvisa. Seção III - Definições Art. 4º Para efeito deste Regulamento Técnico são adotadas as seguintes definições: 278
279 I - advertências e precauções: instruções sobre medidas antecipadas ou avisos que favorecem o uso correto, prudente e seguro do medicamento para prevenir agravos à saúde e que podem indicar a limitação do uso do medicamento, mas que não o contra-indique; II - bula: documento legal sanitário que contém informações técnico-científicas e orientadoras sobre os medicamentos para o seu uso racional; III - bula em formato especial: bula fornecida à pessoa portadora de deficiência visual em formato apropriado para atender suas necessidades. Pode ser disponibilizada: em áudio ou em texto com formato passível de conversão para áudio utilizando um meio magnético (ex: disquetes), meio óptico (ex: CDs, DVDs), meio eletrônico (ex: cartão de memória, pen drive) ou serviços e recursos da internet (ex: correio eletrônico, World Wide Web - www); impressas em Braille ou com fonte ampliada; IV - bula para o paciente: bula destinada ao paciente, aprovada pela Anvisa, com conteúdo sumarizado, em linguagem apropriada e de fácil compreensão; V - bula para o profissional de saúde: bula destinada ao profissional de saúde, aprovada pela Anvisa, com conteúdo detalhado tecnicamente; VI - Bulário Eletrônico: base de dados disponibilizada no Portal da Anvisa que contém as últimas versões aprovadas dos textos de bulas de medicamentos ou outros documentos que possam substituí-las; VII - Bula Padrão: bula definida como padrão de informação para harmonização das bulas de medicamentos específicos, fitoterápicos, genéricos e similares, cujos textos são publicados no Bulário Eletrônico. Para os medicamentos específicos e fitoterápicos, as Bulas Padrão são elaboradas pela Anvisa. Para os medicamentos genéricos e similares, as Bulas Padrão são as bulas dos medicamentos eleitos como medicamentos de referência; VIII - contra-indicação: qualquer condição relativa a uma doença, ao doente ou a uma interação medicamentosa, que impliquea não utilização do medicamento. Caso essa condição não seja observada, poderá acarretar efeitos nocivos graves à saúde do usuário do medicamento ou mesmo levá-lo a óbito; IX - deficiência visual: caracterizada como cegueira, quando a acuidade visual é igual ou menor que 0,05 no melhor olho, com a melhor correção óptica; ou como baixa visão, quando a acuidade visual está entre 0,3 e 0,05 no melhor olho, com a melhor correção óptica; ou nos casos em que a somatória da medida do campo visual em ambos os olhos for igual ou menor que 60 ; ou na ocorrência simultânea de quaisquer das condições anteriores; X - destinação comercial: venda permitida para farmácias e drogarias; XI - destinação hospitalar: venda permitida para hospitais, clínicas e ambulatórios; XII - destinação institucional: venda permitida para os programas governamentais com destino aos postos de dispensação de medicamentos vinculados ao Sistema Único de Saúde; XIII - destinação profissional/ empresa especializada: venda permitida para profissionais ou empresa especializada; XIV - embalagem: invólucro, recipiente ou qualquer forma de acondicionamento removível, ou não, destinado a cobrir, empacotar, envasar, proteger ou manter, especificamente ou não, medicamentos; XV - embalagem hospitalar: embalagem secundária de medicamentos de venda com ou sem exigência de prescrição médica, utilizada para o acondicionamento de medicamentos com destinação hospitalar; XVI - embalagem múltipla: embalagem secundária de medicamentos de venda sem exigência de prescrição médica dispensados exclusivamente nas embalagens primárias; XVII - embalagem primária: embalagem que mantém contato direto com o medicamento; XVIII - embalagem secundária: embalagem externa do produto, que está em contato com a embalagem primária ou envoltório intermediário, podendo conter uma ou mais embalagens primárias; XIX - evento adverso: qualquer ocorrência médica desfavorável, que pode ocorrer durante o tratamento com um medicamento, mas que não possui, necessariamente, relação causal com esse tratamento; 279
280 XX- forma farmacêutica: estado final de apresentação que os princípios ativos farmacêuticos possuem após uma ou mais operações farmacêuticas executadas com a adição de excipientes apropriados ou sem a adição de excipientes, a fim de facilitar a sua utilização e obter o efeito terapêutico desejado, com características apropriadas a uma determinada via de administração; XXI - forma farmacêutica básica: tipo geral da forma farmacêutica (ex: cápsula, comprimido, suspensão, solução) que agrupa formas farmacêuticas específicas com características semelhantes; XXII - forma farmacêutica específica: forma farmacêutica na maioria das vezes originária da forma farmacêutica básica, com a indicação da forma de apresentação e administração e de outras características da formulação (ex: aerossol, para diluição, para infusão). São agrupadas pela forma farmacêutica básica; XXIII - freqüência de reações adversas: proporção da ocorrência de experiência nociva entre os expostos a um dado medicamento que, para efeito de padronização, deve ser referenciada da seguinte forma: muito comum, comum (freqüente), incomum (infreqüente), rara e muito rara; XXIV - gravidade de reações adversas: refere-se ao desfecho de uma reação após o uso do medicamento em um determinado paciente, classificada em graves e não graves. São consideradas graves as situações apresentadas a seguir: óbito; ameaça à vida, quando há risco de morte no momento do evento; hospitalização ou prolongamento de hospitalização já existente, caracterizada como um atendimento hospitalar com necessidade de internação ou um prolongamento da internação devido a um evento adverso; incapacidade significativa ou persistente, quando ocorre uma interrupção substancial da habilidade de uma pessoa conduzir as funções de sua vida normal; anomalia congênita; qualquer suspeita de transmissão de agente infeccioso por meio de um medicamento e evento clinicamente significante, caracterizado como qualquer evento decorrente do uso de medicamentos que ocasione a necessidade de intervenção médica, a fim de se evitar óbito, risco à vida, incapacidade significativa ou hospitalização. Qualquer outro evento que não esteja incluído nos critérios de evento adverso grave é considerado não grave; XXV - Guia de Redação de Bulas: documento publicado no Portal da Anvisa que apresenta alguns princípios de redação clara, concisa e acessível para o leitor de bulas; XXVI - Guia de Submissão Eletrônica de Bulas: documento publicado no Portal da Anvisa que estabelece as orientações para a submissão dos arquivos eletrônicos das bulas dos medicamentos à Anvisa; XXVII - incompatibilidade medicamentosa: interações do tipo físico-químicas que ocorrem fora do organismo durante o preparo e administração dos medicamentos de uso parenteral, inviabilizando a terapêutica clínica. Pode ocorrer entre medicamento-medicamento, medicamento-solução, medicamento-veículo, medicamento-material de embalagem, medicamento-recipiente, medicamento-impureza e freqüentemente resultam no aparecimento de coloração diferente, precipitação ou turvação de uma solução, liberação de gás, formação de espuma ou inativação do princípio ativo; XXVIII - interação medicamentosa: é uma resposta farmacológica ou clínica causada pela interação de medicamento-medicamento, medicamento-alimento, medicamento-substância química, medicamento-exame laboratorial e não laboratorial, medicamento planta medicinal, medicamento-doença cujo resultado final pode ser a alteração dos efeitos desejados ou a ocorrência de eventos adversos; XXVIX - Memento Terapêutico: publicação de responsabilidade dos laboratórios oficiais destinada aos profissionais de saúde que contempla as informações técnico-científicas orientadoras sobre medicamentos disponibilizadas nas bulas dos profissionais de saúde, para a promoção do seu uso racional; XXX - populações especiais: subgrupos de populações que apresentam características especiais, tais como: crianças, idosos, lactentes, gestantes, diabéticos, alérgicos a um ou mais componentes do medicamento, cardiopatas, hepatopatas, renais crônicos, celíacos, imunodeprimidos, atletas e outros que necessitam de atenção especial ao utilizar determinado medicamento; 280
281 XXXI - reação adversa a medicamentos: qualquer resposta a um medicamento que seja prejudicial, não-intencional, e que ocorra nas doses normalmente utilizadas, em seres humanos, para profilaxia, diagnóstico e tratamento de doenças ou para a modificação de uma função fisiológica; XXXII - restrição de uso: limitação de uso de um medicamento quanto à população alvo, podendo ser para uso pediátrico, para uso adulto ou para uso adulto e pediátrico; XXXIII - restrição de prescrição: limitação de prescrição de um medicamento de acordo com a sua categoria de venda, podendo ser de venda sem exigência de prescrição médica, venda sob prescrição médica, com ou sem retenção de receita, de acordo com norma específica; XXXIV - restrição de destinação: limitação do estabelecimento alvo para a venda do medicamento, sendo que uma mesma apresentação pode ter mais de uma destinação, podendo ser comercial, hospitalar, institucional e profissional/ empresa especializada; XXXV - Sistema Braille: processo de leitura e escrita em relevo, com base em 64 (sessenta e quatro) símbolos resultantes da combinação de 6 (seis) pontos, dispostos em duas colunas de 3 (três) pontos; XXXVI - severidade da reação adversa: a intensidade da reação adversa que pode ser classificada como: leve, quando não afeta a atividade cotidiana habitual do paciente; moderada, quando perturba ou altera a atividade cotidiana habitual do paciente; e severa (intensa), quando impede a atividade cotidiana habitual do paciente; XXXVII - Uso restrito a hospitais: medicamentos cuja administração é permitida apenas em ambiente hospitalar, independente da restrição de destinação, definidos em norma específica; e XXXVIII - via de administração: local do organismo por meio do qual o medicamento é administrado. CAPÍTULO II - DA FORMA E CONTEÚDO DAS BULAS Art. 5 Quanto à forma, as bulas dos medicamentos devem: I - apresentar fonte Times New Roman no corpo do texto com tamanho mínimo de 10 pt (dez pontos) nas bulas para o paciente e 8 pt (oito pontos) nas bulas para o profissional de saúde, com espaçamento simples entre letras; II - quando houver necessidade, o limite de redução do espaçamento entre letras será de -10% (menos dez por cento); III - apresentar texto com espaçamento entre linhas de no mínimo 11 pt (onze pontos) nas bulas para o paciente e 9 pt (nove pontos) nas bulas para o profissional de saúde; IV - apresentar colunas de texto com no mínimo 50 mm (cinquenta milímetros) de largura; V - ter o texto alinhado à esquerda ou justificado, hifenizado ou não; VI - utilizar caixa alta e negrito para destacar as perguntas e os itens de bula; VII - possuir texto sublinhado e itálico apenas para nomes científicos; VIII - ser impressas na cor preta em papel branco de forma que, quando a bula estiver sobre uma superfície, a visualização da impressão na outra face não interfira na leitura. 1 Para a impressão de bulas em formato especial, com fonte ampliada, deve ser utilizada a fonte Verdana com tamanho mínimo de 24 pt (vinte e quatro pontos), com o texto corrido e não apresentar colunas. 2 Para a impressão de bulas em formato especial, em Braille, o arranjo dos pontos e o espaçamento entre as celas Braille devem atender às diretrizes da Comissão Brasileira de Braille - CBB e das Normas Brasileiras de Acessibilidade editadas pela Associação Brasileira de Normas Técnicas - ABNT. 3 Para a disponibilização da bula no Bulário Eletrônico e por meio de serviços e recursos de internet, o texto deve ser corrido e não apresentar colunas. Art. 6 Quanto ao conteúdo, as bulas devem contemplar as informações preconizadas no Anexo I desta resolução, seguindo a ordem das partes e itens estabelecida. 281
282 1 As bulas para o paciente devem conter os itens relativos às partes Identificação do Medicamento, Informações ao Paciente e Dizeres Legais e os seus textos devem: I - ser organizados na forma de perguntas e respostas; II - ser claros e objetivos sem a repetição de informações; III - ser escritos em linguagem acessível, com redação clara e concisa, conforme proposto no Guia de Redação de Bulas, de forma a facilitar compreensão do conteúdo pelo paciente; e IV - possuir termos explicativos após os termos técnicos, quando eles forem utilizados e se fizer necessária uma explicação para compreensão do conteúdo pelo paciente. 2 As bulas para o profissional de saúde devem conter os itens relativos às partes Identificação do Medicamento, Informações Técnicas aos Profissionais de Saúde e Dizeres Legais e os seus textos devem: I - ser organizados na forma de itens; II - ser claros e sem a repetição de informações, de forma a facilitar compreensão do conteúdo; e III - contemplar a referência a sinais, sintomas e doenças conforme a terminologia preconizada pela Classificação Internacional de Doenças, dispostas na publicação mais atualizada. 3º É permitido suprimir itens de bula previstos no Anexo I ou partes deles, mediante justificativa técnica sobre sua não aplicabilidade para o medicamento, e realizar nova numeração dos itens, quando necessário. 4 Pode ser exigida a inclusão de outras informações não previstas no caput deste artigo, conforme normas específicas de registro e de notificação de medicamentos. 5 Todos os medicamentos devem possuir bulas para o paciente e bulas para o profissional de saúde, visando sua disponibilização no Bulário Eletrônico, por meio de serviços e recursos de internet e constituição do processo de registro do medicamento na Anvisa, independente do tipo de bula a ser disponibilizado na embalagem. Art. 7 As bulas devem conter apenas as informações relativas às apresentações comercializadas dos medicamentos. Art. 8 As bulas para o paciente devem contemplar informações sobre as apresentações comercializadas com a mesma forma farmacêutica básica e via de administração. 1 Os medicamentos com formas farmacêuticas específicas de liberação modificada devem apresentar bulas distintas, de forma a conferir maior segurança na utilização dos medicamentos. 2 Os medicamentos com formas farmacêuticas básicas e específicas que possuem concentrações com indicações terapêuticas diferentes, devem possuir bulas distintas, de forma a conferir maior segurança na utilização dos medicamentos. 3 Os medicamentos com formas farmacêuticas injetáveis com mesma formulação e diferentes vias de administração podem possuir uma única bula. Art. 9 As bulas para os profissionais de saúde podem contemplar as informações relativas a todas as apresentações comercializadas do medicamento, independente das formas farmacêuticas, vias de administração e concentrações. Art. 10. As frases de advertências a serem inseridas nos textos das bulas devem seguir a redação definida em norma específica. Seção I - Dos medicamentos que não possuem Bula Padrão 282
283 Art. 11. As bulas dos medicamentos que não possuem Bula Padrão devem ser elaboradas pelas empresas para cada produto obedecendo ao disposto nesta resolução, quanto à forma e conteúdo. Parágrafo único. Os medicamentos dinamizados de notificação simplificada devem conter Folheto de Orientação ao Consumidor em substituição à bula, o qual deve apresentar os itens relativos às partes Identificação do Medicamento, Informações ao Paciente e Dizeres Legais, previstos no Anexo I desta resolução, com exceção do item "1. Para quê este medicamento é indicado?", conforme disposto em norma específica. Art. 12. Os medicamentos de notificação simplificada podem ser dispensados da apresentação de bula, sendo esta substituída por rótulo, conforme norma específica. Parágrafo único. Os medicamentos dinamizados de notificação simplificada devem conter Folheto de Orientação ao Consumidor, conforme disposto nesta resolução e em norma específica. Seção II - Dos medicamentos que possuem Bula Padrão Art. 13. As bulas dos medicamentos específicos e fitoterápicos que possuem Bula Padrão publicada no Bulário Eletrônico devem ser harmonizadas com esta e os campos: I - sinalizados com XXX na Bula Padrão e as informações relacionadas ao modo de usar devem ser preenchidos pela empresa de acordo com as características do produto aprovadas no registro; II - sublinhados na Bula Padrão não devem constar das bulas finais disponibilizadas para os medicamentos. Art. 14. As bulas dos medicamentos genéricos e similares devem ser harmonizadas com as suas respectivas Bulas Padrão no tocante à forma e ao conteúdo relativo às informações sobre a eficácia e segurança para uso do medicamento. 1º As bulas dos medicamentos genéricos e similares podem diferir das suas respectivas Bulas Padrão apenas nas informações específicas para cada produto, que devem estar de acordo com as características farmacotécnicas aprovadas no registro, contidas nas partes: I - Identificação do Medicamento, descrita no Anexo I desta resolução, com exceção da informação da via de administração e idade mínima para uso adulto e pediátrico; II - Informações ao Paciente, descritas no Anexo I desta resolução, quanto às frases de advertências específicas relacionadas aos excipientes, aos cuidados de armazenamento, ao prazo de validade, às orientações de preparo e reações adversas que forem relacionadas à formulação do medicamento e não apenas ao princípio ativo; III - Informações aos Profissionais de Saúde, descritas no Anexo I desta resolução, quanto às frases de advertências específicas relacionadas aos excipientes, aos cuidados de armazenamento, ao prazo de validade, às orientações de preparo e às incompatibilidades e reações adversas que forem relacionadas à formulação do medicamento e não apenas ao princípio ativo; IV - Dizeres Legais, descritos no Anexo I desta resolução, com exceção dos dizeres relacionados à restrição de venda ou uso que devem ser os mesmos dispostos nas respectivas Bulas Padrão. 2º As bulas dos medicamentos genéricos e similares devem contemplar apenas as informações das Bulas Padrão relativas às formas farmacêuticas e concentrações para as quais há registros relacionados para os genéricos e similares. 283
284 CAPÍTULO III - DAS ALTERAÇÕES NOS TEXTOS DE BULAS Art. 15. À Anvisa reserva-se o direito de exigir alterações nos textos de bulas, sempre que julgar necessário, por razões técnicocientíficas ou por informações provenientes da farmacovigilância, visando o esclarecimento dos pacientes e profissionais de saúde e a segurança no uso dos medicamentos. Parágrafo único. Poderá ser exigida a inclusão de alerta de segurança, após a parte Identificação do Medicamento, em formato retangular com fundo preto, com os dizeres determinados pelas áreas responsáveis da Anvisa, no prazo a ser estabelecido conforme o risco sanitário. Art. 16. São passíveis de notificação de alteração de bula, com implementação imediata sem manifestação prévia da Anvisa, as atualizações de informações nas bulas a seguir relacionadas: I - à Lista de Denominação Comum Brasileira (DCB); II - ao Vocabulário Controlado; III - ao novo enquadramento dos medicamentos quanto à restrição de uso e prescrição que venha a ser exigida em norma específica; IV - à incorporação de frases de alerta que venha a ser exigida em norma específica; V - aos Dizeres Legais, quanto ao Telefone do Serviço de Atendimento ao Consumidor (SAC), e ao nome do responsável técnico, número de inscrição e sigla do Conselho Regional de Farmácia; VI - aos Dizeres Legais, quanto à razão social das empresas internacionais; e VII - aos Dizeres Legais, quanto à razão social das empresas nacionais, após aprovação da Anvisa da alteração de razão social. Parágrafo único. Para as alterações nos textos de bulas relativas aos incisos deste artigo, as bulas devem ser notificadas, submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, e ser disponibilizadas em até 180 (cento e oitenta) dias após as atualizações, devendo ser implementadas independentemente de manifestação prévia da Anvisa. Seção I - Dos medicamentos que não possuem Bula Padrão Art. 17. Para alterações nos textos de bulas dos medicamentos que não possuem Bula Padrão, relativas às informações sobre a segurança para uso do medicamento, as bulas devem ser peticionadas ou notificadas, e se limitam aos seguintes itens de bulas: I - "QUANDO NÃO DEVO USAR ESTE MEDICAMENTO?"; II - "O QUE DEVO SABER ANTES DE USAR ESTE MEDICAMENTO?"; III - "QUAIS OS MALES QUE ESTE MEDICAMENTO PODE ME CAUSAR?"; IV - "O QUE FAZER SE ALGUÉM USAR UMA GRANDE QUANTIDADE DESTE MEDICAMENTO DE UMA SÓ VEZ?"; V - "CONTRA-INDICAÇÕES"; VI - "ADVERTÊNCIAS E PRECAUÇÕES"; VII - "INTERAÇÕES MEDICAMENTOSAS"; VIII - "REAÇÕES ADVERSAS"; e IX - "SUPERDOSE". 284
285 1 Para a inclusão de informações de segurança, as bulas devem ser notificadas, submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, e ser disponibilizadas em até 180 (cento e oitenta) dias após a notificação, independentemente de manifestação prévia da Anvisa. 2 Para a exclusão ou alteração de informações de segurança, as bulas devem ser peticionadas na Anvisa e, posteriormente à análise, ser submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, e ser disponibilizadas em até 180 (cento e oitenta) dias após a aprovação da petição pela Anvisa. 3 Alterações em outros itens de bula devem estar vinculadas a petições de pós-registro ou de renovação. Art. 18. Para todas as alterações nos textos de bulas dos medicamentos que não possuem Bula Padrão, referentes informações sobre a segurança para uso do medicamento, provocadas pelas empresas matrizes ou autoridades sanitárias dos países que concederam o registro original aos medicamentos, as empresas titulares dos registros no Brasil devem peticionar ou notificar, conforme o caso, a alteração de textos de bulas em até 30 (trinta) dias após ciência. Art. 19. Para as alterações nos textos de bulas dos medicamentos que não possuem Bula Padrão provenientes de petições de pós-registro ou renovação de registro, conforme normas específicas, as bulas devem ser submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, e ser disponibilizadas em até 180 (cento e oitenta) dias após a aprovação da petição pela Anvisa. Parágrafo único. Para as alterações dos rótulos do medicamento de notificação simplificada que substituem as informações de bulas, de acordo com norma específica, seus textos devem ser submetidos eletronicamente à Anvisa, conforme instrução do caput deste artigo. Seção II- Dos medicamentos que possuem Bula Padrão Art. 20. Para as alterações nos textos de bulas dos medicamentos que possuem Bula Padrão, vinculadas às alterações de suas respectivas Bulas Padrão, exceto para as informações específicas do produto, as bulas devem ser notificadas e disponibilizadas em até 180 (cento e oitenta) dias após a publicação das Bulas Padrão no Bulário Eletrônico, devendo ser implementadas, independentemente de manifestação prévia da Anvisa. Art. 21. Para alterações nos textos de bulas dos medicamentos genéricos e similares, provenientes de petições de alterações de pós-registro ou renovação de registros, conforme norma específica, relacionadas às informações específicas para cada produto, as bulas devem ser disponibilizadas em até 180 (cento e oitenta) dias após a aprovação da petição pela Anvisa. Art. 22. Para as alterações nos textos de bulas dos medicamentos específicos e fitoterápicos que possuem Bula Padrão, provenientes de petições de pós-registro ou renovação de registro, conforme norma específica, relacionadas às informações dos campos sinalizados com XXX na Bula Padrão e preenchidos pelas empresas, as bulas devem ser disponibilizadas em até 180 (cento e oitenta) dias após aprovação da petição pela Anvisa. Parágrafo único. Novas informações podem ser incluídas na bula de um determinado medicamento fitoterápico em relação à Bula Padrão e ser inseridas apenas na bula do medicamento testado quando forem provenientes de petições de pós-registro aprovadas, conforme descrito em norma específica, referentes à: 285
286 I - inclusão de nova indicação terapêutica, com comprovação por meio de ensaios pré-clínicos, quando necessário, e clínicos, realizados com o produto, e; II - ampliação de uso, com comprovação do aumento da população alvo do medicamento, advinda de estudos de Fase III e IV. Art. 23. As empresas titulares do registro de medicamentos genéricos e similares que identificarem informações insuficientes sobre a segurança do medicamento em uma Bula Padrão, podem peticionar solicitação de sua revisão, desde que devidamente justificada, cabendo à Anvisa a análise quanto à pertinência da solicitação e verificação da necessidade de tais alterações. 1º Não são passíveis de revisão, por meio da petição prevista no caput deste artigo, as informações específicas para cada produto, previstas nesta resolução, que podem diferir da Bula Padrão para as bulas dos medicamentos genéricos e similares. 2º A deliberação sobre a necessidade de revisão da Bula Padrão será comunicada pela Anvisa à empresa solicitante e à empresa titular do registro do Medicamento de Referência que terá um prazo de até 90 (noventa) dias, conforme o risco sanitário, para peticionar ou notificar a alteração de texto de bula, com a possibilidade de recorrer da decisão em até 10 (dez) dias. Art. 24. As Bulas Padrão de medicamentos fitoterápicos e específicos serão avaliadas e republicadas periodicamente pela Anvisa e as alterações nos textos de Bula Padrão deverão constar nas bulas de todos os medicamentos específicos e fitoterápicos relacionados. Parágrafo único. No caso de surgirem novas informações que devam ser inseridas nas Bulas Padrão de medicamentos fitoterápicos e específicos, qualquer interessado pode enviar sugestões à Anvisa, por meio da Central de Atendimento ou carta à área técnica responsável, cabendo à Anvisa a análise e verificação da pertinência da solicitação e necessidade de implementar as alterações. Art. 25. As alterações nos textos de Bulas Padrão que forem publicadas no Bulário Eletrônico serão divulgadas pela Anvisa por meio de publicação de alertas em seu portal. CAPÍTULO IV - DA DISPONIBILIZAÇÃO DAS BULAS Seção I - Por meio das embalagens dos medicamentos Art. 26. As embalagens dos medicamentos devem conter bulas com conteúdo atualizado no mercado, conforme o Bulário Eletrônico, obedecendo ao estabelecido nesta Resolução, quanto à forma e ao conteúdo. Parágrafo único. Na parte Dizeres Legais das bulas para o paciente e para o profissional de saúde deve constar a data de sua aprovação ou a data de aprovação da Bula Padrão com a qual a bula foi harmonizada e/ou atualizada. Art. 27. As embalagens de medicamentos destinadas aos estabelecimentos que realizam atividade de dispensação de medicamentos para o paciente, prevista na legislação específica, devem conter bula para o paciente. Parágrafo único. É facultativo disponibilizar a bula do paciente nas embalagens dos medicamentos de uso restrito a hospitais, com destinação profissional/empresa especializada ou para administração por profissional de saúde. 286
287 Art. 28. As embalagens dos medicamentos de uso restrito a hospitais, com destinação profissional/empresa especializada ou para administração por profissional de saúde devem conter bula para o profissional de saúde. Art. 29. As embalagens múltiplas, embalagens com destinação hospitalar e embalagens com destinação institucional devem conter um número mínimo de bulas que atenda à quantidade relativa ao menor período de tratamento discriminado na indicação do medicamento. 1 No caso de medicamentos para uso agudo que são dispensados para o paciente na embalagem primária, o número de bulas para o paciente deve ser equivalente ao número de embalagens primárias. 2 No caso de medicamento de uso contínuo que são dispensados para o paciente na embalagem primária, deve-se utilizar como referência o período de 30 dias de tratamento para se calcular o número de bulas para o paciente a serem disponibilizadas na embalagem secundária. 3 No caso de medicamentos uso restrito a hospitais, de uso profissional/empresa especializada ou para administração por profissional de saúde, deve-se dispor de, no mínimo, 1 (uma) bula para o profissional de saúde. 4 As bulas podem ser acondicionadas fora da embalagem secundária. Art. 30. As embalagens dos medicamentos fracionáveis devem conter o número de bulas preconizado em normas específicas. Seção II - Por meio dos Mementos Terapêuticos Art. 31. Os laboratórios oficiais podem disponibilizar as informações para os profissionais de saúde por meio dos Mementos Terapêuticos e sua distribuição deve garantir o acesso à informação para os profissionais de saúde do SUS. Parágrafo único. Caso não haja publicação de Memento Terapêutico, os Laboratórios Oficiais devem disponibilizar bulas para os profissionais de saúde por meio das embalagens dos medicamentos, obedecendo ao disposto nesta resolução quanto à forma e conteúdo. Art. 32. Os Mementos Terapêuticos devem contemplar as bulas para os profissionais de saúde dos medicamentos registrados pelos Laboratórios Oficiais, que devem obedecer ao disposto nesta resolução quanto à forma e conteúdo. Parágrafo único. Em cada bula para o profissional de saúde que constitui o Memento Terapêutico deve constar a data de sua aprovação ou a data de aprovação da Bula Padrão com a qual a bula foi harmonizada e/ou atualizada. Art. 33. A publicação de Memento Terapêutico não isenta os Laboratórios Oficiais da submissão eletrônica, da harmonização e da alteração de textos de bulas, conforme disposto nesta resolução. Seção III - Por meio do Bulário Eletrônico Art. 34. Serão publicadas no Bulário Eletrônico, no Portal Anvisa, as últimas versões dos textos de bulas dos medicamentos para o paciente e para o profissional de saúde, regulamentadas por esta Resolução, e os textos do rótulo do medicamento de notificação simplificada que substituem informação de bula, conforme norma específica. 287
288 1º Somente serão publicados no Bulário Eletrônico as bulas e os textos de rótulos, que substituem informação de bula, referentes aos medicamentos comercializados. 2º A utilização do conteúdo do Bulário Eletrônico é permitida, desde que se façam constar a fonte de onde foram retiradas as informações, qual seja: a empresa titular do registro do medicamento, bem como a data da respectiva consulta, e sejam respeitados os direitos autorais, sem prejuízo de sanções cíveis e criminais em eventuais alterações, que são expressamente proibidas. Seção IV- Por meio de serviços e recursos de internet Art. 35. As empresas podem disponibilizar por meio de serviços e recursos de internet as bulas para o paciente e para o profissional da saúde de todos os seus medicamentos registrados, sem acesso restrito, desde que reproduzam fielmente as últimas versões aprovadas pela Anvisa. 1º Devem ser veiculados, nos serviços e recursos de internet que disponibilizam as bulas, alertas sobre o risco da automedicação ou do uso do medicamento em desacordo com o estabelecido pelo prescritor. 2º Na parte Dizeres Legais das bulas para o paciente e para o profissional de saúde deve constar a data de sua aprovação ou a data de aprovação da Bula Padrão com a qual a bula foi harmonizada e/ou atualizada. 3 As bulas disponibilizadas por meio de serviços e recursos de internet devem ter formato de arquivo passível de conversão em áudio e ampliação de fonte. Seção V - Para pessoas portadoras de deficiência visual Art. 36. As bulas em formato especial devem ser disponibilizadas gratuitamente pelas empresas titulares do registro do medicamento, mediante solicitação da pessoa física portadora de deficiência visual. 1 As empresas devem disponibilizar para escolha da pessoa portadora de deficiência visual bulas: I - em áudio ou em texto com formato passível de conversão para áudio utilizando meio magnético, meio óptico, meio eletrônico ou serviços e recursos da internet; II - impressas em Braille; III - impressas com fonte ampliada. 2 Os textos dos rótulos de medicamentos de notificação simplificada, que substituem a bula, e dos Folhetos de Orientação ao Consumidor, no caso de medicamentos dinamizados, também devem ser disponibilizadas em formato especial, conforme definido neste artigo. Art. 37. A empresa titular de registro do medicamento deve enviar a bula em formato especial solicitado pela pessoa física portadora de deficiência visual no prazo máximo de até 10 (dez) dias úteis após recebimento do pedido. Parágrafo único. A empresa titular de registro do medicamento deve disponibilizar a bula em áudio, por meio do seu Serviço Telefônico de Atendimento ao Consumidor (SAC) ou outro de sua responsabilidade, com a opção de leitura parcial ou total, para escolha da pessoa portadora de deficiência visual e acesso rápido às informações sobre o medicamento. Art. 38. A empresa titular do registro do medicamento tem a responsabilidade de garantir e zelar pela veracidade e atualização das informações prestadas nas bulas em formato especial, objeto desta Resolução. 288
289 Parágrafo único. Na parte Dizeres Legais das bulas para o paciente e para o profissional de saúde deve constar a data de sua aprovação ou a data de aprovação da Bula Padrão com a qual a bula foi harmonizada. Art. 39. A empresa titular de registro do medicamento tem a responsabilidade de arquivar, por 5 (cinco) anos, o registro das solicitações e do envio das bulas em formato especial para pessoas portadoras de deficiência visual, contendo no mínimo as seguintes informações: I - nome completo do requerente; II - endereço residencial completo para correspondência; III - formato de bula solicitada; IV - nome comercial do medicamento; V - a denominação genérica de cada princípio ativo ou insumos ativos, no caso de medicamentos dinamizados, ou nomenclatura botânica, no caso de medicamentos fitoterápicos; VI - concentração e forma farmacêutica; VII - data e comprovante de envio da bula; e VIII - data e comprovante de recebimento da bula. Parágrafo único. As empresas deverão manter em sigilo os dados pessoais do requerente, devendo esses ser utilizados exclusivamente para os fins do atendimento. CAPÍTULO V - DAS DISPOSIÇÕES FINAIS E TRANSITÓRIAS Art. 40. As bulas que sofrerão adequação a esta resolução devem apresentar conteúdo de acordo com a última bula aprovada, sendo permitidas apenas: I - inclusões de informações que passaram a ser exigidas por esta resolução; II - atualizações de informações relativas às alterações de texto de bulas que são passíveis de notificação e que podem ser implementadas independentemente da manifestação prévia da Anvisa, conforme disposto nesta resolução; III - inclusões de informações de segurança; IV- inclusões de informações relacionadas a alterações pósregistro deferidas pela Anvisa após a última bula aprovada. Art. 41. Para os medicamentos já registrados que não possuem Bula Padrão, suas bulas devem ser adequadas quanto à forma e ao conteúdo, obedecendo ao disposto nesta resolução, e ser peticionadas e submetidas eletronicamente à Anvisa, conforme Guia de Submissão Eletrônica de Bulas, no prazo de até: I (cento e oitenta) dias, a partir da publicação desta resolução, para todos os medicamentos registrados sob categorias relacionadas ao aparelho digestivo, metabolismo e nutrição; ao aparelho respiratório; ao aparelho cardiovascular; a parasitoses, neoplasias e infecções; à defesa, imunologia e alergia e aos produtos naturais e homeopáticos, conforme detalhado na Lista 1 no Portal da Anvisa; II (trezentos e sessenta) dias, a partir da publicação desta resolução, para os medicamentos registrados sob categorias relacionadas a sangue e órgãos hematopoiéticos; à pele, mucosas e aparelhos auditivo e visual; ao aparelho geniturinário e hormônios sexuais; ao sistema endócrino, exclusive metabolismo e aparelho genital; ao sistema nervoso; ao sistema musculoesquelético; ao diagnóstico e à situações não especificadas, conforme detalhado na Lista 2 no Portal da Anvisa. 1 Os medicamentos que forem incluídos na Lista de Medicamento de Referência durante o período de adequação a esta resolução passam a ter suas bulas enquadradas como Bula Padrão que devem ser adequadas quanto à forma e 289
290 ao conteúdo, obedecendo ao disposto nesta resolução, e serem peticionadas e submetidas eletronicamente à Anvisa, conforme Guia de Submissão Eletrônica de Bulas nos prazos estabelecidos nos incisos deste artigo ou em até 180 (cento e oitenta) dias, a partir da sua inclusão na Lista de Medicamento de Referência, valendo o maior prazo. 2 Para os medicamentos que forem incluídos na Lista de Medicamento de Referência, as suas bulas já adequadas a esta resolução devem ser submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, em até 30 (trinta) dias a partir de sua inclusão na Lista de Medicamento de Referência, caso não estejam publicadas no Bulário Eletrônico. 3 Os medicamentos específicos e fitoterápicos, que não possuem Bula Padrão elaborada e publicada pela Anvisa, devem seguir as instruções do caput deste artigo. Art. 42. Para a solicitação de registro de medicamentos que não possuem Bula Padrão, suas bulas devem ser elaboradas pelas empresas, obedecendo ao disposto nesta resolução quanto à forma e conteúdo, e ser submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, em até 30 (trinta) dias após o início da comercialização. Art. 43. Para os medicamentos já registrados que possuem Bula Padrão, suas bulas devem ser harmonizadas com a Bula Padrão, obedecendo ao disposto nesta resolução, e ser notificadas em até 90 (noventa) dias a partir da publicação das suas respectivas Bulas Padrão no Bulário Eletrônico da Anvisa. Art. 44. Para a solicitação de registro de medicamentos que possuem Bula Padrão, suas bulas devem ser harmonizadas com a Bula Padrão, obedecendo ao disposto nesta resolução. 1 Para os medicamentos genéricos e similares, cuja Bula Padrão não estiver adequada quanto à forma e ao conteúdo, obedecendo ao disposto nesta resolução, suas bulas devem seguir a última bula aprovada do medicamento de referência. 2 Para os medicamentos específicos ou fitoterápicos, cuja Bula Padrão não estiver adequada quanto à forma e ao conteúdo, obedecendo ao disposto nesta resolução, suas bulas devem seguir a última bula elaborada e publicada pela Anvisa. Art. 45. Para os medicamentos de notificação simplificada já aprovados, suas bulas ou Folhetos de Orientação ao Consumidor devem ser adequadas quanto à forma e ao conteúdo, obedecendo ao disposto nesta resolução, e ser notificadas e submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, em até 180 (cento e oitenta) dias a partir da publicação desta resolução. Parágrafo único. Os textos dos rótulos dos medicamentos de notificação simplificada que substituem as informações de bulas, de acordo com norma específica, devem ser submetidos eletronicamente à Anvisa, conforme instruções do caput deste artigo. Art. 46. Para a solicitação de notificação simplificada de medicamentos, suas bulas ou Folhetos de Orientação ao Consumidor devem ser elaborados pelas empresas, obedecendo ao disposto nesta resolução, e ser submetidas eletronicamente à Anvisa, conforme instruções do Guia de Submissão Eletrônica de Bulas, em até 30 (trinta) dias após o início da comercialização. Parágrafo único. Os textos dos rótulos dos medicamentos de notificação simplificada que substituem as informações de bulas, de acordo com norma específica, devem ser submetidos eletronicamente à Anvisa, conforme instrução do caput deste artigo. 290
291 Art. 47. As bulas adequadas a esta resolução, quanto à forma e conteúdo, devem ser disponibilizadas por meio das embalagens dos medicamentos, dos Mementos Terapêuticos, se for o caso, e para as pessoas portadoras de deficiência visual, conforme previsto nesta resolução, em até: I (cento e oitenta) dias após a publicação da bula no Bulário Eletrônico, para os medicamentos que não possuem Bula Padrão, sendo este o tempo previsto para o esgotamento do estoque; e, II (cento e oitenta) dias a partir da publicação da Bula Padrão no Bulário Eletrônico, para os medicamentos que possuem Bula Padrão, independente da manifestação prévia da Anvisa quanto à notificação de alteração do texto de bula para adequação a esta resolução, sendo este o tempo previsto para o esgotamento do estoque. Parágrafo único. A empresa titular do registro deve disponibilizar as últimas versões aprovadas pela Anvisa das bulas dos medicamentos, por meio do correio eletrônico ou da sua leitura, parcial ou total, pelo Serviço Telefônico de Atendimento ao Consumidor (SAC), conforme escolha e necessidade das pessoas portadoras de deficiência visual, em até 30 (trinta) dias após publicação desta resolução, mesmo que as bulas ainda não estejam adequadas a esta norma, de forma a garantir às pessoas portadoras de deficiência visual acesso às informações constantes nas bulas durante o prazo de adequação à norma. Art. 48. Após as alterações de texto de bula, provenientes de petições ou notificações, as empresas devem disponibilizar as novas bulas, nos prazos previstos nesta resolução, por meio das embalagens dos medicamentos, dos Mementos Terapêuticos, se for o caso, dos serviços e recursos de internet, se utilizados pela empresa, e para as pessoas portadoras de deficiência visual, sendo estes prazos os previstos para esgotamento de estoque. Art. 49. Compete à autoridade de vigilância sanitária estadual, municipal e federal proceder, nas inspeções rotineiras nas indústrias farmacêuticas ou importadoras de medicamentos, a verificação das alterações nos textos de bula, em consonância com as datas de fabricação dos lotes, datas de publicação da bula no Bulário Eletrônico da Anvisa e prazos para adequação estabelecidos nesta resolução. Art. 50. O descumprimento das disposições contidas nesta resolução e no regulamento por ela aprovado constituem infração sanitária, nos termos da Lei n 6.437, de 20 de agosto de 1977, sem prejuízo das responsabilidades civil, administrativa e penal cabíveis. Art. 51. Fica revogada a Portaria SVS/MS nº 110, de 10 de março de 1997; as Resoluções da Diretoria Colegiada RDC nº 140, de 29 de maio de 2003, RDC nº 126, de 16 de maio de 2005, Resolução RDC nº 94, de 11 de dezembro de 2008, e RDC n. 95 de 11 de dezembro de 2008; o item 10.1 da parte III do anexo I da RDC nº 16, de 02 de março de 2007; o item h.1 da parte II do anexo da RDC nº 17, de 02 de março de 2007; e o anexo IV da RDC n 26, de 30 março de Art. 52. Esta Resolução entra em vigor na data de sua publicação. ANEXO I - IDENTIFICAÇÃO DO MEDICAMENTO: Citar o nome comercial do medicamento. Citar a denominação genérica do(s) princípio(s) ativo(s), utilizando a Denominação Comum Brasileira (DCB). 291
292 Para medicamentos fitoterápicos, informar espécie vegetal (Gênero + epíteto específico) para sua a denominação genérica, a família, a parte da planta utilizada e a nomenclatura popular. No caso de medicamentos dinamizados, descrever os insumos utilizando a nomenclatura das farmacopéias e compêndios reconhecidos pela Anvisa para sua a denominação genérica. Para medicamentos fitoterápicos, registrados com base na tradicionalidade de uso, inserir as frases: "Medicamento fitoterápico registrado com base no uso tradicional." (em negrito) "Não é recomendado o uso por período prolongado enquanto estudos clínicos amplos sobre sua segurança não forem realizados." Para medicamentos dinamizados, incluir a frase, conforme a categoria do medicamento, em negrito: "Medicamento Homeopático", "Medicamento Antroposófico" ou "Medicamento Anti-homotóxico". Para os medicamentos genéricos, incluir a frase "Medicamento Genérico, Lei nº 9.787, de 1999". APRESENTAÇÕES Citar apresentações comercializadas, informando:- a forma farmacêutica;- a concentração do(s) princípio(s) ativo(s), por unidade de medida ou unidade farmacotécnica, conforme o caso; - a quantidade total de peso, volume líquido ou unidades farmacotécnicas, conforme o caso;- a quantidade total de acessórios dosadores que acompanha as apresentações, quando aplicável. Citar via de administração, usando caixa alta e negrito. Incluir a frase, em caixa alta e em negrito, "USO ADULTO", "USO ADULTO E PEDIÁTRICO ACIMA DE " ou "USO PEDIÁTRICO ACIMA DE ", indicando a idade mínima, em meses ou anos, para qual foi aprovada no registro o uso do medicamento. No caso de medicamentos sem restrição de uso por idade, conforme aprovado no registro, incluir a frase "USO ADULTO e PEDIÁTRICO". COMPOSIÇÃO Para o(s) princípio(s) ativo(s), descrever a composição qualitativa, conforme DCB, e quantitativa e indicar equivalência sal-base, quando aplicável. Para os excipientes, descrever a composição qualitativa, conforme DCB. Para formas farmacêuticas líquidas, quando o solvente for alcoólico, mencionar a graduação alcoólica do produto final. Para medicamentos com forma farmacêutica líquida e em gotas, informar a equivalência de gotas para cada mililitro (gotas/ml) e massa por gota (mg/gotas). Para os medicamentos para Terapia de Reidratação Oral (TRO), informar a quantidade dos princípios ativos em unidades de massa ou massa/volume, e na forma de meq/l. Para os medicamentos injetáveis classificados como soluções parenterais de grande volume (SPGV), informar a composição qualitativa e quantitativa, percentual, conteúdo eletrolítico em meq/l ou mmol/l e osmolaridade. Para medicamentos fitoterápicos, a composição do medicamento deve indicar a relação real, em peso ou volume, do derivado vegetal utilizado a correspondência em marcadores e a descrição do derivado. 292
293 Para medicamentos dinamizados, descrever a composição qualitativa e quantitativa para os insumos ativos, informando a potência e escala de cada insumo, e a composição qualitativa para os insumos inertes. INFORMAÇÕES AO PACIENTE: 1. PARA QUE ESTE MEDICAMENTO É INDICADO? Descrever as indicações de uso do medicamento devidamente registradas na Anvisa indicando o objetivo terapêutico, ou seja, se é destinado para o tratamento, diagnóstico, auxiliar no diagnóstico ou prevenção. Exemplos: Este medicamento é destinado ao tratamento de... Este medicamento é destinado ao tratamento e prevenção de... Para medicamentos dinamizados, descrever sucintamente em qual(is) situação(ções) clínica(s) o medicamento se propõe a agir. Destacar que: Este medicamento é um auxiliar no tratamento de... Para medicamentos dinamizados, inserir a seguinte frase, em negrito: "A indicação deste medicamento somente poderá ser alterada a critério do prescritor." 2. COMO ESTE MEDICAMENTO FUNCIONA?Descrever resumidamente as ações do medicamento em linguagem acessível à população em geral. Informar o tempo médio estimado para início da ação terapêutica do medicamento, quando aplicável. 3. QUANDO NÃO DEVO USAR ESTE MEDICAMENTO? Descrever as contra-indicações para o uso do medicamento. No caso de contra-indicação de uso do medicamento para populações especiais, incluir as seguintes frases, em negrito: "Este medicamento é contra-indicado para uso por " (informando a população especial)."este medicamento é contra-indicado para menores de " (citando a idade em meses ou anos). No caso de contra-indicação de uso do medicamento por homens ou mulheres, incluir uma das seguintes frases, em negrito: " Este medicamento é contra-indicado para uso por homens." ou "Este medicamento é contra-indicado para uso por mulheres." No caso de contra-indicação do uso do medicamento por mulheres grávidas, incluir, em negrito, de acordo com o período gestacional, as frases de alerta associadas às categorias de risco de fármacos destinados às mulheres grávidas, conforme norma específica. No caso de contra-indicação para o uso de princípios ativos, classe terapêutica e excipientes, incluir, em negrito, as frases de alerta previstas em norma específica. Para medicamentos dinamizados, descrever, quando houver, as contra-indicações específicas ou fatores que limitem a utilização do medicamento, como hipersensibilidade aos insumos ativos (obrigatoriamente para dinamizações 1CH, 2DH ou menor) e insumos inertes. 4. O QUE DEVO SABER ANTES DE USAR ESTE MEDICAMENTO? 293
294 Descrever as advertências e precauções para o uso adequado do medicamento, incluindo, quando aplicável, informações sobre: - cuidados e advertências para populações especiais; - alterações de condições fisiológicas, incluindo aquelas que possam afetar a capacidade de dirigir veículos e operar máquinas; e - sensibilidade cruzada. No caso de medicamentos destinados ao tratamento de doenças infecto-contagiosas, inserir orientações sobre as medidas de higiene recomendadas em cada caso. Nos casos de advertências e precauções para uso do medicamento por mulheres grávidas, incluir, em negrito, de acordo com o período gestacional, as frases de alerta associadas às categorias de risco de fármacos destinados às mulheres grávidas, conforme norma específica. No caso de advertências e precauções para o uso de princípios ativos, classe terapêutica e excipientes, incluir, em negrito, as frases de alerta previstas em norma específica. Para medicamentos que podem causar doping, conforme especificação do Comitê Olímpico Internacional - COI, incluir a seguinte frase, em negrito: "Este medicamento pode causar doping." Para medicamentos dinamizados, incluir, em negrito, as frases de advertências e precauções relativas aos insumos inertes, conforme o caso: "Este medicamento contém ÁLCOOL."; "Este medicamento contém LACTOSE."; "Atenção diabéticos: este medicamento contém SACAROSE." Descrever as interações medicamentosas, por potencial de significância clínica, esclarecendo quanto às conseqüências e prejuízos para o paciente ou para o tratamento, agrupando os casos similares e dispondo informações, quando aplicável, sobre:- as interações medicamento-medicamento, inclusive com medicamentos fitoterápicos. Caso a interação seja relacionada a uma classe terapêutica, exemplificar com os princípios ativos mais importantes;- as interações medicamento-planta medicinal; - as interações medicamento-substância química, com destaque para o álcool e nicotina; - as interações medicamento-exame laboratorial e não laboratorial; - as interações medicamentos-doenças, caso não estejam dispostas juntamente com contra-indicações, advertências e precauções; - as interações medicamento- alimento. Incluir a frase, em negrito: "Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento." Para os medicamentos vendidos sob prescrição médica, incluir a seguinte frase, em negrito: 294
295 "Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde." 5. ONDE, COMO E POR QUANTO TEMPO POSSO GUARDAR ESTE MEDICAMENTO? Descrever os cuidados de conservação do medicamento. Incluir as seguintes frases, em negrito: "Número de lote e datas de fabricação e validade: vide embalagem." "Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original." Descrever os cuidados específicos de conservação para medicamentos que uma vez abertos ou preparados para o uso sofram redução do prazo de validade original ou alteração do cuidado de conservação original, incluindo uma das seguintes frases, em negrito:" Após aberto, válido por " (indicando o tempo de validade após aberto, conforme estudos de estabilidade do medicamento) "Após preparo, manter por " (indicando o cuidado de conservação e o tempo de validade após preparo, conforme estudos de estabilidade do medicamento) Descrever as características físicas e organolépticas do produto e outras características do medicamento, inclusive após a reconstituição e/ou diluição. Incluir as seguintes frases, em negrito:"antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo." Incluir a seguinte frase, em negrito:"todo medicamento deve ser mantido fora do alcance das crianças." Para medicamentos dinamizados, inserir a seguinte frase, em negrito: "Proteger da luz solar e de fontes de radiação eletromagnética, como por exemplo: forno de microondas, aparelho celular, televisão, etc." 6. COMO DEVO USAR ESTE MEDICAMENTO? Descrever as principais orientações sobre o modo correto de preparo, manuseio e aplicação do medicamento. Incluir o risco de uso por via de administração não recomendada, quando aplicável. Para soluções para diluição ou pós ou granulados para solução, suspensão ou emulsão de uso oral ou injetável, incluir: - o procedimento detalhado para reconstituição e/ou diluição antes da administração; - o(s) diluente(s) a ser(em) utilizado(s); - o volume final do medicamento preparado; e - concentração do medicamento preparado. Descrever a posologia, incluindo as seguintes informações: - dose para forma farmacêutica e concentração, expresso, quando aplicável, em unidades de medida ou unidade farmacotécnica correspondente em função ao tempo, definindo o intervalo de administração em unidade de tempo;- a dose inicial e de manutenção, quando aplicável; - duração de tratamento; 295
296 - vias de administração;- orientações para cada indicação terapêutica nos casos de posologias distintas; - orientações para uso adulto e/ou uso pediátrico, de acordo com o aprovado no registro; e- orientações sobre o monitoramento e ajuste de dose para populações especiais. Para os medicamentos com apresentação líquida para uso sistêmico, expressar a dose do medicamento em unidade de medida, em massa ou Unidade Internacional (UI) do princípio ativo, por quilograma (kg) corpóreo ou superfície corporal. Para as formas farmacêuticas de liberação modificada expressar a dose liberada por unidade de tempo e tempo total de liberação do princípio ativo. Descrever o limite máximo diário de administração do medicamento expresso em unidades de medida ou unidade farmacotécnica correspondente. Para medicamento dinamizado, citar a dose máxima diária quando o insumo ativo for considerado tóxico (tabela constante da Farmacopéia Homeopática dos Estados Unidos - HPUS) e a dinamização for tal que possa induzir efeitos tóxicos se utilizado além do limite estabelecido. Para os medicamentos vendidos sob prescrição médica, incluir as seguintes frases, em negrito: "Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico." Para os medicamentos isentos de prescrição médica, incluir a seguinte frase, em negrito: "Siga corretamente o modo de usar. Em caso de dúvidas sobre este medicamento, procure orientação do farmacêutico. Não desaparecendo os sintomas, procure orientação de seu médico ou cirurgião- dentista." Conforme característica da forma farmacêutica, incluir a seguinte frase, em negrito: "Este medicamento não deve ser partido, aberto ou mastigado." (para comprimidos revestidos, cápsulas e compridos de liberação modificada e outras que couber) ou "Este medicamento não deve ser cortado." (para adesivos e outras que couber) Para medicamentos dinamizados, alertar para o aparecimento de sintomas novos ou agravação de sintomas atuais, quando aplicável, e incluir as seguintes frases, em negrito: "Informe ao seu médico, cirurgião-dentista o aparecimento de sintomas novos, agravação de sintomas atuais ou retorno de sintomas antigos." "O uso inadequado do medicamento pode mascarar ou agravar sintomas." "Consulte um clínico regularmente. Ele avaliará corretamente a evolução do tratamento. Siga corretamente suas orientações." 7. O QUE DEVO FAZER QUANDO EU ME ESQUECER DE USAR ESTE MEDICAMENTO? Descrever a conduta necessária, caso haja esquecimento de administração (dose omitida), quando for o caso. Orientar sobre a atitude adequada quando houver a possibilidade de síndrome de abstinência. Incluir a seguinte frase, em negrito: "Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista." 296
297 Para os medicamentos dinamizados, inserir a seguinte frase, em negrito: "Caso você esqueça de usar o medicamento, não duplique a quantidade de medicamento na próxima tomada." 8. QUAIS OS MALES QUE ESTE MEDICAMENTO PODE ME CAUSAR? Citar as reações adversas, ordenando-as e agrupando-as por freqüência, das mais comuns para as muitas raras, explicitando os sinais e sintomas relacionados a cada uma. Informar sobre a gravidade e severidade, quando aplicável. Incluir, quando possível, os seguintes textos informativos e explicativos sobre a incidência de ocorrência das reações adversas, antes de citá-las: "Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento):." "Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento):." "Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento):." "Reação rara (ocorre entre 0,01% e 0,1% dos pacientes que utilizam este medicamento):." "Reação muito rara (ocorre em menos de 0,01% dos pacientes que utilizam este medicamento):." Ao classificar a freqüência das reações, utilizar os seguintes parâmetros: Freqüência das Reações Adversas Parâmetros > 1/10 (> 10%) muito comum > 1/100 e 1/10 (> 1% e 10%) comum (freqüente) > 1/1.000 e 1/100 (> 0,1% e 1%) incomum (infreqüente) > 1/ e 1/1.000 (> 0,01% e 0,1%) rara 1/ ( 0,01%) muito rara Incluir as seguintes frases, em negrito:"informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento." Substituir a frase anterior pela seguinte, quando se tratar de um medicamento novo, referente à molécula nova isolada ou em associação, no Brasil, em condições normais de comercialização ou dispensação durante os cinco primeiros anos de comercialização: "Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, informe seu médico ou cirurgião- dentista." Substituir a frase anterior pela seguinte, quando já houver passado o prazo dos cinco primeiros anos para molécula nova isolada ou em associação, e incluí-la durante cinco anos de comercialização do medicamento com nova indicação terapêutica, nova via de administração, nova concentração, nova forma farmacêutica e/ou nova associação no país: "Atenção: este produto é um medicamento que possui no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos 297
298 imprevisíveis ou desconhecidos. Nesse caso, informe seu médico". (incluindo no espaço: nova indicação terapêutica, nova via de administração, nova concentração, nova forma farmacêutica e/ou nova associação, conforme o caso) Para medicamentos dinamizados, quando couber, informar quais os tipos mais comuns e freqüência das possíveis agravações do medicamento, obrigatoriamente somente para aqueles dentro da faixa de toxicidade (com tarja) nas dinamizações 1CH, 2DH ou menor. Para medicamentos dinamizados, inserir a frase: "Em caso de sintomas que causem mal estar durante o tratamento, procure seu médico ou farmacêutico." 9. O QUE FAZER SE ALGUÉM USAR UMA QUANTIDADE MAIOR DO QUE A INDICADA DESTE MEDICAMENTO? Descrever os sintomas que caracterizam a superdose e orientar quanto a medidas preventivas que amenizem o dano até a obtenção do socorro médico. Para medicamentos dinamizados, incluir a conduta adequada para atendimento emergencial, especialmente para medicamentos que contenham insumos ativos nas dinamizações 1CH, 2DH ou menor, conforme o caso. Inserir as seguintes frases, em negrito:"em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para , se você precisar de mais orientações." INFORMAÇÕES TÉCNICAS AOS PROFISSIONAIS DE SAÚDE 1. INDICAÇÕES Descrever as indicações de uso do medicamento devidamente registradas na Anvisa informando o objetivo terapêutico, ou seja, se é destinado para o tratamento, diagnóstico, auxiliar no diagnóstico ou profilaxia. Exemplos: Este medicamento é destinado ao tratamento de... Este medicamento é destinado ao tratamento e profilaxia de? Para medicamentos dinamizados, descrever sucintamente em qual(is) situação(ções) clínica(s) o medicamento se propõe a agir. Destacar que: Este medicamento é um auxiliar no tratamento de RESULTADOS DE EFICÁCIA Apresentar o resultado de eficácia do grupo tratado com o medicamento em questão e o grupo controle, incluindo diferenças que permitam uma maior visualização da relevância do tratamento e citando as referências bibliográficas. Para os medicamentos genéricos e similares, apresentar os resultados de eficácia do seu respectivo medicamento de referência, mencionando apenas o(s) nome(s) do(s) princípio(s) ativo(s). Para medicamentos específicos, fitoterápicos e dinamizados, apresentar os resultados de eficácia quando aplicável. 3. CARACTERÍSTICAS FARMACOLÓGICAS Descrever o medicamento com as suas propriedades farmacológicas, tanto as farmacodinâmicas quanto as farmacocinéticas, fundamentadas técnico-cientificamente. Informar o tempo médio estimado para início da ação terapêutica do medicamento, quando aplicável Para medicamentos específicos, fitoterápicos e dinamizados, apresentar as características farmacológicas quando aplicável. 298
299 Para medicamentos dinamizados, descrever o medicamento com as suas propriedades fundamentadas técnicocientificamente no âmbito da terapêutica homeopática, antroposófica ou homotoxicológica, conforme o caso. 4. CONTRA-INDICAÇÕES Descrever as contra-indicações para o uso do medicamento. No caso de contra-indicação de uso do medicamento para populações especiais, incluir as seguintes frases, em negrito:"este medicamento é contra-indicado para uso por." (informando a população especial)."este medicamento é contra-indicado para menores de." (citando a idade em meses ou anos). No caso de contra-indicação de uso do medicamento por homens ou mulheres, incluir um das seguintes frases, em negrito:"este medicamento é contra-indicado para uso por homens." ou "Este medicamento é contra-indicado para uso por mulheres." No caso de contra-indicação do uso do medicamento por mulheres grávidas, indicar e descrever a categoria de risco na gravidez, de acordo com período gestacional, e incluir, em negrito, as frases de alerta associadas às categorias de risco de fármacos destinados às mulheres grávidas, conforme norma especifica. No caso de contra-indicação para o uso de princípios ativos, classe terapêutica e excipientes, incluir, em negrito, as frases de alerta previstas em norma específica. Para medicamentos dinamizados, descrever, quando houver, as contra-indicações específicas ou fatores que limitem a utilização do medicamento, como hipersensibilidade aos insumos ativos (obrigatoriamente para dinamizações 1CH, 2DH ou menor) e insumos inertes. 5. ADVERTÊNCIAS E PRECAUÇÕES Descrever as advertências e precauções para o uso adequado do medicamento. Incluir, quando aplicável, informações sobre:- cuidados e advertências para populações especiais;- alterações de condições fisiológicas, incluindo aquelas que possam afetar a capacidade de dirigir veículos e operar máquinas;- sensibilidade cruzada; e- teratogenicidade, mutagenicidade e reprodução, quando houver, e outros cuidados necessários. No caso de medicamentos destinados ao tratamento de doenças infecto-contagiosas, inserir orientações sobre as medidas de higiene recomendadas em cada caso. Nos casos de advertências e precauções para uso do medicamento por mulheres grávidas, indicar e descrever a categoria de risco na gravidez, de acordo com período gestacional, e incluir, em negrito, as frases de alerta associadas às categorias de risco de fármacos destinados às mulheres grávidas, conforme norma específica. No caso de advertências e precauções para o uso de princípios ativos, classe terapêutica e excipientes, incluir, em negrito, as frases de alerta previstas em norma específica. Para medicamentos que podem causar doping, conforme especificação do Comitê Olímpico Internacional - COI, incluir a seguinte frase, em negrito: "Este medicamento pode causar doping. 299
300 "Para medicamentos dinamizados, incluir, em negrito, as frases de advertências e precauções relativas aos insumos inertes, conforme o caso: "Este medicamento contém ÁLCOOL."; "Este medicamento contém LACTOSE."; "Atenção diabéticos: este medicamento contém SACAROSE." 6. INTERAÇÕES MEDICAMENTOSAS Descrever as interações medicamentosas, por potencial de significância clínica, esclarecendo quanto às conseqüências e prejuízos para o paciente ou para o tratamento, agrupando os casos similares e dispondo informações, quando aplicável, sobre:- as interações medicamento-medicamento, inclusive com medicamentos fitoterápicos. Caso a interação seja relacionada a uma classe terapêutica, exemplificar com os princípios ativos mais importantes. - as interações medicamento-planta medicinal; - as interações medicamento-substância química, com destaque para o álcool e nicotina; - as interações medicamento-exame laboratorial e não laboratorial; - as interações medicamentos-doenças, caso não estejam dispostas juntamente com contra-indicações, advertências e precauções; e - as interações medicamento- alimento. 7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO Descrever os cuidados específicos para o armazenamento do medicamento e informar o prazo de validade do medicamento a partir da data de fabricação, aprovado no registro, citando o número de meses. Incluir as seguintes frases, em negrito: "Número de lote e datas de fabricação e validade: vide embalagem." "Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original."descrever os cuidados específicos de conservação para medicamentos que uma vez abertos ou preparados para o uso sofram redução do prazo de validade original ou alteração do cuidado de conservação original, incluindo uma das seguintes frases, em negrito:"após aberto, válido por " (indicando o tempo de validade após aberto, conforme estudos de estabilidade do medicamento) "Após preparo, manter por " (indicando o cuidado de conservação e o tempo de validade após preparo, conforme estudos de estabilidade do medicamento) Descrever as características físicas e organolépticas do produto e outras características do medicamento, inclusive após a reconstituição e/ou diluição. Incluir as seguintes frases, em negrito:"antes de usar, observe o aspecto do medicamento." Incluir a seguinte expressão em negrito:"todo medicamento deve ser mantido fora do alcance das crianças" Para medicamentos dinamizados, inserir a seguinte frase, em negrito:" Proteger da luz solar e de fontes de radiação eletromagnética, como por exemplo: forno de microondas, aparelho celular, televisão, etc." 300
301 8. POSOLOGIA E MODO DE USAR Descrever as principais orientações sobre o modo correto de preparo, manuseio e aplicação do medicamento. Incluir o risco de uso por via de administração não recomendada, quando aplicável. Para soluções para diluição ou pós ou granulados para solução, suspensão ou emulsão de uso oral ou injetável, incluir: - o procedimento detalhado para reconstituição e/ou diluição antes da administração; - o(s) diluente(s) a ser(em) utilizado(s); - o volume final do medicamento preparado; - concentração do medicamento preparado. Para soluções de uso parenteral, incluir informações sobre incompatibilidade esclarecendo as consequências e possíveis prejuízos para o tratamento. Descrever a posologia, incluindo as seguintes informações: - dose para forma farmacêutica e concentração, expresso, quando aplicável, em unidades de medida ou unidade farmacotécnica correspondente em função ao tempo, definindo o intervalo de administração em unidade de tempo; - a dose inicial e de manutenção, quando aplicável; - intervalos de administração (em minutos ou horas); - duração de tratamento; - vias de administração; - orientações para cada indicação terapêutica nos casos de posologias distintas; - orientações para uso adulto e/ou uso pediátrico, de acordo com o aprovado no registro; - orientações sobre o monitoramento e ajuste de dose para populações especiais. Para os medicamentos com apresentação líquida para uso sistêmico, expressar a dose do medicamento em unidade de medida, em massa ou Unidade Internacional (UI) do princípio ativo, por quilograma (kg) corpóreo ou superfície corporal. Para as formas farmacêuticas de liberação modificada expressar a dose liberada por unidade de tempo e tempo total de liberação do princípio ativo. Descrever o limite máximo diário de administração do medicamento expresso em unidades de medida ou unidade farmacotécnica correspondente. Para medicamento dinamizado, citar a dose máxima diária quando o insumo ativo for considerado tóxico (tabela constante da Farmacopéia Homeopática dos Estados Unidos - HPUS) e a dinamização for tal que possa induzir efeitos tóxicos se utilizado além do limite estabelecido. Conforme característica da forma farmacêutica, incluir a seguinte frase, em negrito: "Este medicamento não deve ser partido, aberto ou mastigado." (para comprimidos revestidos, cápsulas e compridos de liberação modificada e outras que couber) "Este medicamento não deve ser cortado." (para adesivos e outras que couber) 9. REAÇÕES ADVERSAS 301
302 Citar as reações adversas, ordenando-as e agrupando-as por freqüência, das mais comuns para as muitas raras, explicitando os sinais e sintomas relacionados a cada uma. Informar sobre a gravidade e severidade, quando aplicável. Incluir, quando possível, os seguintes textos informativos e explicativos sobre a incidência de ocorrência das reações adversas, antes de citá-las: "Reação muito comum (> 1/10):." "Reação comum (> 1/100 e 1/10):." "Reação incomum (> 1/1.000 e 1/100):." "Reação rara (> 1/ e 1.000):." "Reação muito rara ( 1/10.000):." Ao classificar a freqüência das reações, utilizar os seguintes parâmetros: Freqüência das Reações Adversas Parâmetros > 1/10 (> 10%) muito comum > 1/100 e 1/10 (> 1% e 10%) comum (freqüente) > 1/1.000 e 1/100 (> 0,1% e 1%) incomum (infreqüente) > 1/ e 1/1.000 (> 0,01% e 0,1%) rara 1/ ( 0,01%) muito rara Inserir a seguinte frase: "Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em ou para a Vigilância Sanitária Estadual ou Municipal." (incluindo no espaço o endereço eletrônico atualizado do NOTIVISA) Substituir a frase anterior pela seguinte, quando se tratar de um medicamento novo, referente à molécula nova isolada ou em associação, no Brasil, em condições normais de comercialização ou dispensação durante os cinco primeiros anos de comercialização: "Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária? NOTIVISA, disponível em ou para a Vigilância Sanitária Estadual ou Municipal." (incluindo no espaço o endereço eletrônico atualizado do NOTIVISA) Substituir a frase anterior pela seguinte, quando já houver passado o prazo dos cinco primeiros anos para molécula nova, isolada ou em associação, e incluí-la durante cinco anos de comercialização do medicamento com nova indicação terapêutica, nova via de administração, nova concentração, nova forma farmacêutica e/ou nova associação no país: "Atenção: este produto é um medicamento que possui no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em ou para a Vigilância Sanitária Estadual ou 302
303 Municipal." (incluindo no primeiro espaço o termo: nova indicação terapêutica, nova via de administração, nova concentração, nova forma farmacêutica e/ou nova associação, conforme o caso; e no último espaço, o endereço eletrônico atualizado do NOTIVISA) Para medicamentos dinamizados, quando aplicável, informar quais os tipos mais comuns e freqüência das possíveis agravações do medicamento, obrigatoriamente somente para aqueles dentro da faixa de toxicidade (com tarja) nas dinamizações 1CH, 2DH ou menor. 10. SUPERDOSE Descrever os sintomas que caracterizam a superdose e orientar quanto a medidas preventivas que amenizem o dano até a obtenção do socorro médico. Para medicamentos dinamizados, incluir a conduta adequada para atendimento emergencial, especialmente para medicamentos que contenham insumos ativos nas dinamizações 1CH, 2DH ou menor, conforme o caso. Inserir a seguinte frase em negrito: "Em caso de intoxicação ligue para , se você precisar de mais orientações." DIZERES LEGAIS Informar a sigla "MS" mais o número de registro no Ministério da Saúde conforme publicado em Diário Oficial da União (D.O.U.), sendo necessários os 9 (nove) dígitos iniciais. Informar o nome, número de inscrição e sigla do Conselho Regional de Farmácia do responsável técnico da empresa titular do registro. Informar o nome e endereço da empresa titular do registro no Brasil. Informar o número do Cadastro Nacional de Pessoa Jurídica (CNPJ) do titular do registro. Inserir a expressão "Indústria Brasileira", quando aplicável. Informar o telefone do Serviço de Atendimento ao Consumidor (SAC), de responsabilidade da empresa titular do registro. Informar o nome e endereço da empresa fabricante, quando ela diferir da empresa titular do registro, citando a cidade e o estado precedidos pela frase "Fabricado por:" e inserindo a frase "Registrado por:" antes dos dados da detentora do registro. Informar o nome e endereço da empresa fabricante, quando o medicamento for importado, citando a cidade e o país precedidos pela frase "Fabricado por" e inserindo a frase "Importado por:" antes dos dados da empresa titular do registro. Informar o nome e endereço da empresa responsável pela embalagem do medicamento, quando ela diferir da empresa titular do registro ou fabricante, citando a cidade e o estado ou, se estrangeira, a cidade e o país, precedidos pela frase "Embalado por:" e inserindo a frase "Registrado por:" ou "Importando por:", conforme o caso, antes dos dados da empresa titular do registro; Informar, se descrito na embalagem do medicamento, o nome e endereço da empresa responsável pela comercialização do medicamento, citando a cidade e o estado precedidos pela frase "Comercializado por" e incluindo a frase "Registrado por:" antes dos dados da detentora do registro. É facultativo incluir a logomarca da empresa farmacêutica titular do registro, bem como das empresas fabricantes e responsáveis pela embalagem e comercialização do medicamento, desde que não prejudiquem a presença das informações obrigatórias e estas empresas estejam devidamente identificadas nos dizeres legais. Incluir as seguintes frases, quando for o caso: 303
304 "Uso restrito a hospitais" (para os medicamentos de uso restrito a hospitais); "Venda sob prescrição médica" (para os medicamentos de venda sob prescrição médica); "Siga corretamente o modo de usar, não desaparecendo os sintomas procure orientação médica" (para os medicamentos vendidos sem exigência de prescrição médica); "Uso sob prescrição médica." (para embalagens com destinação institucional); " Venda proibida ao comércio." (para os medicamentos com destinação institucional). Incluir as frases de restrições de venda, uso e dispensação previstas na norma específica para produtos controlados. Incluir, exceto nos textos de bula a serem submetidos eletronicamente à Anvisa, uma das seguintes frases, conforme o caso, em negrito: "Esta bula foi aprovada pela Anvisa em (dia/mês/ano)" (informando a data de publicação da bula no Bulário Eletrônico) "Esta bula foi atualizada conforme Bula Padrão aprovada pela Anvisa em (dia/mês/ano)" (informando a data de publicação da respectiva Bula Padrão no Bulário Eletrônico com a qual a bula foi harmonizada e/ou atualizada). Incluir símbolo da reciclagem de papel. 304
305 RDC Nº 71, DE 22 DE DEZEMBRO DE 2009 (NÃO APLICÁVEL A MEDICAMENTOS NOTIFICADOS) Estabelece regras para a rotulagem de medicamentos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária - Anvisa, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, e tendo em vista o disposto no inciso II e nos 1º e 3º do art. 54 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 354 da Anvisa, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada 16 em dezembro de 2009; considerando que a saúde é direito de todos e dever do Estado, garantido mediante políticas sociais e econômicas que visem à redução do risco de doença e de outros agravos e ao acesso universal e igualitário às ações e serviços para sua promoção, proteção e recuperação, nos termos do art. 196 da Constituição da República Federativa do Brasil, de 5 de outubro de 1988; considerando a Lei nº 6.360, de 23 de setembro de 1976, e o Decreto nº , de 5 de janeiro de 1977, que dispõe sobre o sistema de vigilância sanitária a que ficam sujeitos os medicamentos; considerando a Lei nº 9.787, de 10 de fevereiro de 1999, que altera a Lei nº 6.360, de 23 de setembro de 1976, que dispõe sobre o medicamento genérico e sobre a utilização de nomes genéricos em produtos farmacêuticos; considerando a Lei nº 5.991, de 17 de dezembro de 1973, e o Decreto nº , de 10 de junho de 1974 que dispõe sobre ocontrole sanitário do comércio de drogas, medicamentos, insumos farmacêuticos e correlatos; considerando a Lei nº 6.437, de 20 de agosto de 1977, que dispõe sobre as infrações à legislação sanitária federal e estabelece as respectivas penalidades; considerando o direito à informação, às pessoas assistidas, sobre sua saúde conforme previsto nos termos do inciso V do art. 7º da Lei Orgânica da Saúde (LOS), Lei nº 8.080, de 19 de setembro de 1990; considerando o direito à informação adequada e clara sobre os diferentes produtos e serviços, com especificação correta de quantidade, características, composição, qualidade e preço, bem como sobre os riscos que apresentem, conforme o previsto pelo inciso III do art. 6 do Código de Defesa do Consumidor, Lei nº 8078, de 11 de setembro de 1990; considerando que compete à União cuidar da saúde e assistência pública, da proteção e garantia das pessoas portadoras de deficiência, nos termos do inciso II do art. 23 da Constituição; considerando as disposições previstas pela Lei n , de 8 de novembro de 2000, que dá prioridade de atendimento às pessoas que especifica, e dá outras providências; considerando as disposições previstas pela Lei n , de 19 de dezembro de 2000, que estabelece normas gerais e critérios básicos para a promoção da acessibilidade das pessoas portadoras de deficiência ou com mobilidade reduzida, e dá outras providências; 305
306 considerando as disposições previstas pelo Decreto nº 5.296, de 02 de dezembro de 2004, que Regulamenta a Lei n , de 8 de novembro de 2000, que dá prioridade de atendimento às pessoas que especifica, e a Lei n , de 19 de dezembro de 2000, que estabelece normas gerais e critérios básicos para a promoção da acessibilidade das pessoas portadoras de deficiência ou com mobilidade reduzida, e dá outras providências; considerando as diretrizes estabelecidas pela Comissão Brasileira de Braille - CBB, e pela Associação Brasileira de Normas Técnicas - ABNT, quanto a utilização do sistema Braille; considerando o documento Standard Rules on the Equalization of opportunities for person with disabilities adotado pela Assembléia Geral das Organizações das Nações Unidas; considerando as diretrizes, as prioridades e as responsabilidades estabelecidas na Política Nacional de Medicamentos, instituída pela Portaria n /MS/GM, de 30 de outubro de 1998, e aprovada pelo CNS pela Resolução n. 338, de 20 maio de 2004, que busca garantir condições para segurança e qualidade dos medicamentos utilizados no país, promover o uso racional e o acesso da população àqueles considerados essenciais; considerando a importância do acesso à informação imparcial e de qualidade para orientar o autocuidado e a automedicação disposta no Report of the 4th WHO - Consultative Group on the Role of the Pharmacist; considerando que as informações sobre medicamentos devem orientar pacientes e profissionais de saúde, favorecendo o uso racional, os rótulos de medicamentos devem conter informações que permitam identificá-lo, armazená-lo e rastreá-lo adequadamente, além de informar sobre riscos sanitários para algumas populações especiais e dispor que outras informações para o uso seguro do medicamento estarão dispostas na sua bula; considerando as disposições específicas da Resolução RDC n. 59, de 24 de novembro de 2009 que dispõe sobre a implantação do Sistema Nacional de Controle de Medicamentos e definição dos mecanismos para rastreamento de medicamentos, por meio de tecnologia de captura, armazenamento e transmissão eletrônica de dados e dá outras providências; considerando a competência da Anvisa, no cumprimento de suas atribuições regulamentares, quanto a implementação de ações para agilizar a operacionalização de suas atividades administrativas quanto ao registro, atualização e revalidação de produtos; considerando a Medida Provisória no , de 23 de agosto de 2001 que instituiu a isenção do recolhimento de taxa para acréscimo ou alteração de registro, referente aos rótulos de medicamentos; adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação: Art. 1º Fica aprovado o Regulamento Técnico que estabelece as diretrizes para a rotulagem de medicamentos. CAPÍTULO I - DAS DISPOSIÇÕES INICIAIS Seção I - Objetivo 306
307 Art. 2º Este Regulamento possui o objetivo de aprimorar a forma e o conteúdo dos rótulos de todos os medicamentos registrados e comercializados no Brasil, visando garantir o acesso à informação segura e adequada em prol do uso racional de medicamentos. Seção II- Abrangência Art. 3º Este Regulamento se aplica a todos os medicamentos registrados na Anvisa. Seção III- Definições Art. 4º Para efeito deste Regulamento Técnico são adotadas as seguintes definições: I - bula: documento legal sanitário que contém informações técnico-científicas e orientadoras sobre os medicamentos para o seu uso racional; II - destinação comercial: venda permitida para farmácias e drogarias; III - destinação hospitalar: venda permitida para hospitais, clínicas e ambulatórios; IV - destinação institucional: venda permitida para os programas governamentais com destino aos postos de dispensação de medicamentos vinculados ao Sistema Único de Saúde; V - destinação profissional/ empresa especializada: venda permitida para profissionais ou empresa especializada; VI - embalagem: invólucro, recipiente ou qualquer forma de acondicionamento removível, ou não, destinado a cobrir, empacotar, envasar, proteger ou manter, especificamente ou não, medicamentos; VII - embalagem de transporte: embalagem utilizada para transporte de medicamentos acondicionados em suas embalagens primárias ou secundárias; VIII - embalagem hospitalar: embalagem secundária de medicamentos de venda com ou sem exigência de prescrição médica, utilizada para o acondicionamento de medicamentos com destinação hospitalar; IX - embalagem múltipla: embalagem secundária de medicamentos de venda sem exigência de prescrição médica dispensados exclusivamente nas embalagens primárias; X - embalagem primária: embalagem que mantém contato direto com o medicamento; XI - embalagem secundária: embalagem externa do produto, que está em contato com a embalagem primária ou envoltório intermediário, podendo conter uma ou mais embalagens primárias; XII - envoltório intermediário: embalagem opcional que está em contato com a embalagem primária e constitui um envoltório ou qualquer outra forma de proteção removível, podendo conter uma ou mais embalagens primárias, conforme aprovação da Anvisa; XIII - restrição de uso: limitação de uso de um medicamento quanto à população alvo, podendo ser para uso pediátrico, para uso adulto ou para uso adulto e pediátrico; XIV - restrição de prescrição: limitação de prescrição de um medicamento de acordo com a sua categoria de venda, podendo ser de venda sem exigência de prescrição médica, venda sob prescrição médica, com ou sem retenção de receita, de acordo com norma específica; V - restrição de destinação: limitação do estabelecimento alvo para a venda do medicamento, sendo que uma mesma apresentação pode ter mais de uma destinação, podendo ser comercial, hospitalar, institucional e profissional/ empresa especializada; XVI - rótulo: identificação impressa ou litografada, bem como dizeres pintados ou gravados a fogo, pressão ou decalco, aplicados diretamente sobre recipientes, vasilhames, invólucros, envoltórios ou qualquer outro protetor de embalagem; 307
308 XVII - Sistema Braille: processo de leitura e escrita em relevo, com base em 64 (sessenta e quatro) símbolos resultantes da combinação de 6 (seis) pontos, dispostos em duas colunas de 3 (três) pontos; e, XVIII - Uso restrito a hospitais: medicamentos cuja administração é permitida apenas em ambiente hospitalar, independentemente da restrição de destinação, definidos em norma específica. CAPÍTULO II - DAS DISPOSIÇÕES GERAIS PARA OS RÓTULOS DE MEDICAMENTOS Seção I - Das informações para as embalagens secundárias Art. 5 Os rótulos das embalagens secundárias de medicamentos devem conter as seguintes informações: I - o nome comercial do medicamento; II - a denominação genérica de cada princípio ativo, em letras minúsculas, utilizando a Denominação Comum Brasileira (DCB); III - a concentração de cada princípio ativo, por unidade de medida ou unidade farmacotécnica, conforme o caso; IV - a via de administração; V - a quantidade total de peso líquido, volume e unidades farmacotécnicas, conforme o caso; VI - a quantidade total de acessórios dosadores que acompanha as apresentações, quando aplicável; VII - a forma farmacêutica; VIII - a restrição de uso por faixa etária, na face principal, incluindo a frase, em caixa alta, "USO ADULTO", "USO ADULTO E PEDIÁTRICO ACIMA DE ", "USO PEDIÁTRICO ACIMA DE ", indicando a idade mínima, em meses ou anos, para qual foi aprovada no registro o uso do medicamento, ou "USO ADULTO e PEDIÁTRICO", no caso de medicamentos sem restrição de uso por idade, conforme aprovado no registro; IX - a composição qualitativa, conforme Denominação Comum Brasileira (DCB), e quantitativa de cada princípio ativo, incluindo, quando aplicável, a equivalência sal base; X - os cuidados de conservação, indicando a faixa de temperatura e condições de armazenamento, conforme estudo de estabilidade do medicamento; XI - o nome e endereço da empresa titular do registro no Brasil; XII - o nome e endereço da empresa fabricante, quando ela diferir da empresa titular do registro, citando a cidade e o estado, precedidos pela frase "Fabricado por:" e inserindo a frase "Registrado por:" antes dos dados da empresa titular do registro; XIII - o nome e endereço da empresa fabricante, quando o medicamento for importado, citando a cidade e o país precedidos pela frase "Fabricado por" e inserindo a frase "Importado por:" antes dos dados da empresa titular do registro; XIV - o nome e endereço da empresa responsável pela embalagem do medicamento, quando ela diferir da empresa titular do registro ou fabricante, citando a cidade e o estado ou, se estrangeira, a cidade e o país, precedidos pela frase "Embalado por:" e inserindo a frase "Registrado por:" ou "Importando por:", conforme o caso, antes dos dados da empresa titular do registro; XV - o número do Cadastro Nacional de Pessoa Jurídica (CNPJ) do titular do registro; XVI - a expressão "Indústria Brasileira", quando aplicável; XVII - o nome do responsável técnico, número de inscrição e sigla do Conselho Regional de Farmácia da empresa titular do registro; XVIII - telefone do Serviço de Atendimento ao Consumidor (SAC) da empresa titular do registro ou de sua responsabilidade; e, 308
309 XIX - a sigla "MS" adicionada ao número de registro no Ministério da Saúde conforme publicado em Diário Oficial da União (DOU), sendo necessários os treze dígitos. 1 No caso de medicamento genérico e imunoterápico, é proibido usar nome comercial, devendo ser adotada apenas a denominação genérica. 2 No caso de medicamentos injetáveis classificados como Soluções Parenterais de Pequeno Volume (SPPV), tais como solução de cloreto de sódio, água para injeção, solução de glicose e outros açúcares e eletrólitos, é facultativo usar nome comercial, podendo ser adotada apenas a denominação genérica. 3 No caso de medicamentos fitoterápicos, deve-se utilizar a nomenclatura botânica, indicando espécie (Gênero + epíteto específico) para sua a denominação genérica; a concentração de cada princípio ativo deve ser expressa pela concentração de cada derivado vegetal e a composição do medicamento deve indicar a relação real, em peso ou volume, do derivado vegetal utilizado a correspondência em marcadores e a descrição do derivado. 4 No caso de medicamentos dinamizados, deve-se descrever os insumos utilizando a nomenclatura das farmacopéias e compêndios reconhecidos pela Anvisa para sua a denominação genérica e a concentração de cada principio ativo deve ser expressa pela potência e escala de cada insumo ativo. 5 É facultativo incluir a composição qualitativa dos excipientes, conforme Denominação Comum Brasileira (DCB), ou dos insumos inertes, no caso dos medicamentos dinamizados. 6 É facultativo incluir informações sobre a empresa responsável pela comercialização do medicamento, informando o seu nome e endereço, citando a cidade e o estado precedidos pela frase "Comercializado por" e incluindo a frase "Registrado por:" antes dos dados da detentora do registro e informar 7 É permitido incluir a logomarca da empresa farmacêutica titular do registro, bem como das empresas fabricantes e responsáveis pela embalagem e comercialização do medicamento, desde que não prejudiquem a presença das informações obrigatórias. Art. 6 Nos rótulos das embalagens secundárias de medicamentos devem ser inseridas as seguintes frases de advertência: I - "TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS", em caixa alta; e, II - "Informações ao paciente, indicações, contra-indicações e precauções: vide bula" ou "Informações ao profissional de saúde, indicações, contra-indicações e precauções: vide bula", conforme o tipo de bula disponibilizada na embalagem do medicamento, de acordo com norma específica. Art. 7 No caso de contra-indicação, precaução ou advertência para o uso de princípios ativos, classe terapêutica e excipientes, devem-se incluir, em negrito, as frases de advertências previstas em norma específica. Seção II - Das informações para as embalagens primárias Art. 8 Os rótulos das embalagens primárias de medicamentos devem conter as seguintes informações: I - o nome comercial do medicamento; II - a denominação genérica de cada princípio ativo, em letras minúsculas, utilizando a Denominação Comum Brasileira (DCB); III - a concentração de cada princípio ativo, por unidade de medida ou unidade farmacotécnica, conforme o caso; IV - a via de administração; 309
310 V - o nome da titular do registro ou sua logomarca desde que a mesma contenha o nome da empresa; e, VI - o telefone do Serviço de Atendimento ao Consumidor (SAC), da empresa titular do registro ou de sua responsabilidade. 1 No caso de medicamento genérico e imunoterápico, é proibido usar nome comercial, devendo ser adotada apenas a denominação genérica. 2 No caso de medicamentos injetáveis classificados como Soluções Parenterais de Pequeno Volume (SPPV), tais como solução de cloreto de sódio, água para injeção, solução de glicose e outros açúcares e eletrólitos, é facultativo usar nome comercial, podendo ser adotada apenas a denominação genérica. 3 No caso de medicamentos fitoterápicos, deve-se utilizar a nomenclatura botânica, indicando espécie (Gênero + epíteto específico) para sua a denominação genérica e a concentração de cada princípio ativo deve ser expressa pela concentração de cada derivado vegetal. 4 No caso de medicamentos dinamizados, deve-se descrever os insumos utilizando a nomenclatura das farmacopéias e compêndios reconhecidos pela Anvisa para sua a denominação genérica e a concentração de cada principio ativo deve ser expressa pela potência e escala de cada insumo ativo. 5º É permitido incluir o nome ou as logomarcas das empresas responsáveis pela fabricação, embalagem e comercialização dos medicamentos, desde que a mesma contenha o nome da empresa e seja informada a etapa da cadeia de sua responsabilidade, incluindo as frases: "Fabricado por:", "Comercializado por"; "Embalado por", e não se prejudique a legibilidade das informações exigidas para a embalagem primária. 6º É permitido incluir as demais informações previstas para a embalagem secundária na embalagem primária, desde que não prejudiquem a legibilidade das informações obrigatórias. Art. 9 A impossibilidade de fazer constar na embalagem primária todas as informações exigidas nesta Resolução, deve ser justificada à Anvisa no momento da notificação, do registro ou pós registro. Seção III - Das informações para as caixas de transporte Art. 10. Os rótulos das caixas de transporte de medicamentos devem conter, impressas ou etiquetadas, as seguintes informações mínimas: I - o nome comercial do medicamento; II - a denominação genérica de cada princípio ativo, em letras minúsculas, utilizando a Denominação Comum Brasileira (DCB); III - a concentração de cada princípio ativo, por unidade de medida ou unidade farmacotécnica, conforme o caso; IV - a forma farmacêutica; V - o nome da titular do registro ou sua logomarca desde que a mesma contenha o nome da empresa; VI - os cuidados de conservação, indicando a faixa de temperatura e condições de armazenamento, conforme estudo de estabilidade do medicamento. 1 No caso de medicamento genérico e imunoterápico, é proibido usar nome comercial, devendo ser adotada apenas a denominação genérica. 310
311 2 No caso de medicamentos injetáveis classificados como Soluções Parenterais de Pequeno Volume (SPPV), tais como solução de cloreto de sódio, água para injeção, solução de glicose e outros açúcares e eletrólitos, é facultativo usar nome comercial, podendo ser adotada apenas a denominação genérica. 3 No caso de medicamentos fitoterápicos, deve-se utilizar a nomenclatura botânica, indicando espécie (Gênero + epíteto específico) para sua a denominação genérica e a concentração de cada princípio ativo deve ser expressa pela concentração de cada derivado vegetal. 4 No caso de medicamentos dinamizados, deve-se descrever os insumos utilizando a nomenclatura das farmacopéias e compêndios reconhecidos pela Anvisa para sua a denominação genérica e a concentração de cada principio ativo deve ser expressa pela potência e escala de cada insumo ativo. Seção IV - Da disposição das informações nos rótulos Art. 11. As letras utilizadas nos rótulos para identificação do nome comercial do medicamento e para a denominação genérica dos princípios ativos, devem ser de fácil leitura e ostentar o mesmo destaque. Art. 12. A denominação genérica de cada princípio ativo deve ser disposta nos rótulos imediatamente abaixo do nome comercial, respeitando as seguintes regras de proporcionalidade: I - para os medicamentos com até dois princípios ativos, o nome genérico de cada um deve ser disposto com tamanho mínimo de 50% da altura do maior caractere do nome comercial; II - para os medicamentos com três ou mais princípios ativos, o nome genérico de cada um deve ser disposto com tamanho mínimo de 30% da altura do maior caractere do nome comercial. 1 No caso de limitação no campo de impressão para descrever os princípios ativos conforme Denominação Comum Brasileira (DCB), englobando o nome do sal e da base, deve constar no rótulo o nome da substância base com tamanho mínimo de 50% da altura do maior caractere do nome comercial e, imediatamente após, o nome do sal, com tamanho mínimo de 30% da altura do maior caractere da base. 2 No caso de limitação no campo de impressão para descrever os três ou mais princípios ativos, deve constar no rótulo a denominação genérica do princípio ativo que melhor justifique a indicação terapêutica do produto seguida da frase "+ ASSOCIAÇÃO", com tamanho mínimo de 50% da altura do maior caractere do nome comercial e a composição do medicamento, qualitativa e quantitativa de todos os princípios ativos deve estar disposta no rótulo, em uma das faces da embalagem secundária ou, na sua ausência, na embalagem primária. 3 No caso de limitação no campo de impressão para descrever todos os princípios ativos dos polivitamínicos, poliminerais e poliaminoácidos, podem ser adotadas as palavras: Polivitamínico, Polimineral e Poliaminoácido, como denominação genérica, com tamanho mínimo de 50% da altura do maior caractere do nome comercial, e a composição do medicamento, qualitativa e quantitativa de todos os princípios ativos deve estar disposta no rótulo, em uma das faces da embalagem secundária ou, na sua ausência, na embalagem primária. 4 No caso de limitação no campo de impressão para utilizar a denominação genérica do princípio ativo de produtos biológicos, ela pode ser abreviada conforme aprovado no registro. Art. 13. A concentração por unidade de medida ou unidade farmacotécnica de cada princípio ativo que for disposto na identificação do medicamento, deve estar no mesmo campo de impressão, abaixo ou ao lado, do nome comercial ou da denominação genérica dos princípios ativos, com tamanho mínimo de 50% da altura do maior caractere do nome comercial. 311
312 Parágrafo único. Para medicamentos genéricos, a concentração deve estar disposta abaixo da denominação genérica dos princípios ativos com tamanho mínimo de 50% da altura do maior caractere da denominação genérica. Art. 14. A descrição da forma farmacêutica pode ser disposta com a quantidade total de peso líquido, volume ou unidades farmacotécnicas do medicamento. Art. 15. As impressões do nome comercial, denominação genérica de cada princípio ativo e respectivas concentrações, devem ser repetidas nos rótulos das embalagens primárias destrutíveis, com mais de uma dose, visando permitir a identificação do medicamento durante todo o tratamento. Art. 16. No caso de serem incluídas as logomarcas das empresas farmacêuticas: titular do registro, fabricante e responsáveis pela comercialização e embalagem do medicamento, elas devem ter dimensão máxima de 50% do tamanho do nome comercial ou, na sua ausência, da denominação genérica dos princípios ativos. Art. 17. Não podem constar nos rótulos dos medicamentos, designações, símbolos, figuras, representações gráficas ou quaisquer indicações que possam tornar a informação falsa e incorreta, que possibilitem interpretação falsa, equívoco, erro e confusão em relação à verdadeira natureza, composição, procedência, qualidade, forma de uso, finalidade e características do medicamento. 1 É proibido: I - incluir imagens de pessoas fazendo uso do medicamento; II - incluir selos, marcas nominativas, figurativas ou mistas de instituições governamentais, entidades filantrópicas, fundações, associações e sociedades médicas, organizações não-governamentais, associações que representem os interesses dos consumidores ou dos profissionais de saúde e selos de certificação de qualidade, exceto se exigidos em normas específicas; III - inclusão de imagens ou figuras que remetam à indicação do sabor do medicamento; IV - usar expressões ou imagens que possam sugerir que a saúde de uma pessoa poderá ser afetada por não usar o medicamento; e, V - utilizar rótulos com layout semelhante ao de um medicamento com o mesmo princípio ativo, forma farmacêutica e concentração,registrado anteriormente por outra empresa. 2 É permitido: I - utilizar figuras anatômicas, a fim de orientar o profissional de saúde ou o paciente sobre a correta utilização do produto; e, II - informar o sabor do medicamento. Art. 18. É permitido incluir em outro idioma as mesmas informações exigidas para os rótulos de medicamentos, desde que não prejudiquem a legibilidade das informações obrigatórias e estejam de acordo com as informações constantes do registro do medicamento. Seção V - Das informações e dispositivos para rastreabilidade do medicamento 312
313 Art. 19. O número do lote, data de fabricação (mês/ano) e data de validade (mês/ano), devem ser impressos nas embalagens de medicamentos de forma facilmente compreensível, legível e indelével, utilizando letras com a maior dimensão possível para a sua fácil leitura e identificação. 1 A legibilidade destas informações deve ser garantida sem a utilização de instrumentos ópticos, a não ser para aquelas pessoas que necessitem de correção visual. 2 Nas embalagens secundárias é proibido usar exclusivamente de relevo negativo ou positivo, sem cor ou com cor que não mantenha nítido e permanente o contraste com a cor do suporte para a impressão das informações exigidas no caput deste artigo. 3 É facultativo imprimir nas embalagens primárias a data de fabricação (mês/ano). Art. 20. As embalagens secundárias devem conter lacre ou selo de segurança que seja irrecuperável após seu rompimento e permita detectar qualquer tentativa de rompimento, para garantir a inviolabilidade das embalagens. 1º Quando utilizado a colagem de abas, ela deve garantir os requisitos descritos no caput deste artigo para ser considerada um lacre de segurança. 2º Quando utilizado selos de segurança, além das características descritas no caput deste artigo, eles não podem permitir a recolagem e devem conter a identificação personalizada do laboratório. 3º No caso de embalagens que permitam o acesso às embalagens primárias por mais de uma extremidade, ambas devem atender aos requisitos contidos no caput deste artigo. 4 Quando o medicamento for disponibilizado exclusivamente em embalagem primária e for passível de abertura, ela deverá conter lacre ou selo de segurança, conforme características do caput deste artigo. Art. 21. As embalagens de medicamentos devem conter mecanismos de identificação e segurança que possibilitem o rastreamento do produto desde a fabricação até o momento da dispensação, conforme dispostos em normas específicas. Art. 22. É facultativo incluir nas embalagens secundárias de medicamentos ou, na sua ausência, nas embalagens primárias, o código de barras GTIN de identificação do produto, caso elas contenham mecanismos de identificação e segurança que possibilitem o rastreamento do produto desde a fabricação até o momento da dispensação. Parágrafo único. É permitido colocar o Código de Barras GTIN na face lateral da embalagem, sobre a faixa de restrição de prescrição, estruturando uma abertura na mesma. Art. 23. É facultativo incluir nas embalagens secundárias de medicamentos ou, na sua ausência, nas embalagens primárias, a tinta reativa e sob a mesma a palavra "Qualidade" e a logomarca da empresa titular do registro caso elas contenham mecanismos de identificação e segurança que possibilitem o rastreamento do produto desde a fabricação até o momento da dispensação. 1 A tinta reativa deve ser disposta em uma das laterais, na altura da faixa vermelha ou preta, sendo para isto permitido abrir uma janela nas referidas faixas que permita a fixação da tinta. 2 Os medicamentos sem exigência de prescrição médica devem colocar a tinta reativa na altura do local que corresponde à faixa de restrição de uso. 3 Qualquer outro local da face externa da embalagem pode ser utilizado desde que seja justificado tecnicamente, não afete as demais exigências legais e seja colocada uma indicação ao consumidor do local onde se deve raspar. Seção VI - Das informações para as pessoas portadoras de deficiência visual 313
314 Art. 24. As embalagens secundárias de medicamentos que são dispensados para o paciente devem conter em sistema Braille, sem afetar a legibilidade das informações, o nome comercial do medicamento ou, na sua falta, a denominação genérica de cada princípio ativo pela Denominação Comum Brasileira (DCB). 1 No caso de medicamentos fitoterápicos, deve-se utilizar a nomenclatura botânica, indicando espécie (Gênero + epíteto específico). 2 No caso de medicamentos dinamizados, deve-se descrever cada insumo ativo utilizando a nomenclatura das farmacopéias e compêndios reconhecidos pela Anvisa. 3 No caso de medicamentos com mais de quatro princípios ativos, pode-se informar o nome do princípio ativo que justifique a indicação terapêutica do produto seguida da expressão "+ associação". 4 No caso de medicamentos identificados pela denominação genérica de cada princípio ativo, em que haja limitação no campo de impressão para o sistema Braille, pode-se utilizar apenas o nome da base do princípio ativo. CAPÍTULO III - DAS DISPOSIÇÕES ESPECÍFICAS PARA OS RÓTULOS DE MEDICAMENTOS Seção I - Dos medicamentos de venda sob prescrição médica Art. 25. Os rótulos das embalagens secundárias dos medicamentos com venda sob prescrição médica devem possuir faixa vermelha, em toda a sua extensão, no seu terço médio inferior e com largura não inferior a um quinto da maior face. Parágrafo único. É proibido colocar as faixas no rodapé das embalagens, devendo-se respeitar o limite mínimo de 10 mm nas bases das embalagens ou na extremidade contrária a sua abertura. Art. 26. Na faixa vermelha deve ser utilizada a referência de cor vermelha PANTONE 485C, que pode ser obtida através da mistura de pigmentos de qualquer fabricante de tintas, com variações máximas e mínimas aceitáveis para este tom, e ser aplicado um verniz sobre ela. 1 É proibida a utilização de cores nos rótulos de medicamentos que possam causar confusão ou erro na identificação da faixa vermelha. 2 É permitido utilizar o PANTONE 485C fora da faixa vermelha apenas na: I - descrição da concentração; II - descrição da quantidade do medicamento; III - descrição da via de administração; IV - frase "Amostra Grátis", seja nas letras ou em fundo vermelho; V - frase "Nova Fórmula"; e, VI - frase "Agite antes de usar". Art. 27. No interior da faixa vermelha dos medicamentos de venda sob prescrição médica deve ser incluída apenas a frase, em caixa alta, "VENDA SOB PRESCRIÇÃO MÉDICA". Parágrafo único. É permitida a inscrição qualitativa de todos os excipientes na face lateral da embalagem, sobre a faixa vermelha, estruturando uma abertura na mesma, utilizando letras com a maior dimensão possível para a sua fácil leitura e identificação. 314
315 Art. 28. Os rótulos das embalagens primárias dos medicamentos com venda sob prescrição médica devem possuir a frase, em caixa alta, "VENDA SOB PRESCRIÇÃO MÉDICA". Seção II - Dos medicamentos de venda sem exigência de prescrição médica Art. 29. Os rótulos das embalagens secundárias dos medicamentos com venda sem exigência de prescrição médica, além das informações mínimas exigidas nesta Resolução, devem conter: I - a frase, em negrito: "Siga corretamente o modo de usar, não desaparecendo os sintomas procure orientação médica"; II - a indicação do medicamento, conforme disposto para o princípio ativo e classe terapêutica em norma específica; e, III - as contra-indicações de uso do medicamento. Art. 30. Os rótulos das embalagens primárias dos medicamentos sem exigência de prescrição médica, disponibilizados exclusivamente em embalagem primária, além das informações exigidas nesta Resolução, devem possuir: I - a frase "EXIJA A BULA", em caixa alta, com tamanho mínimo de 30% da altura do maior caractere do nome comercial ou, na sua ausência, da denominação genérica; II - a sigla "MS" adicionada ao número de registro no Ministério da Saúde, conforme publicado em Diário Oficial da União (DOU), sendo necessários os treze dígitos; e, III - a restrição de uso por faixa etária, incluindo a frase, em caixa alta, "USO ADULTO", "USO ADULTO E PEDIÁTRICO ACIMA DE ", "USO PEDIÁTRICO ACIMA DE ", indicando a idade mínima, em meses ou anos, para qual foi aprovada no registro o uso do medicamento, ou "USO ADULTO e PEDIÁTRICO", no caso de medicamentos sem restrição de uso por idade, conforme aprovado no registro. Seção III - Dos medicamentos à base de substâncias sujeitas a controle especial Art. 31. Os rótulos das embalagens secundárias dos medicamentos à base de substâncias sujeitas a controle especial devem possuir uma faixa em toda sua extensão, no seu terço médio inferior e na cor vermelha ou preta, conforme definido em norma específica e suas atualizações, para a substância ou lista à qual pertence. Parágrafo único. É proibida a colocação das faixas no rodapé das embalagens, devendo-se respeitar o limite mínimo de 10 mm nas bases das embalagens ou na extremidade contrária sua abertura. Art. 32. Na faixa preta, deve ser utilizada a referência de cor preta PANTONE PROCESSO BLACK C, que pode ser obtida através da mistura de pigmentos de qualquer fabricante de tintas, com variações máximas e mínimas aceitáveis para este tom, e ser aplicado um verniz sobre ela. 1 A faixa preta deve ter largura não inferior a um terço da maior face e exclui a exigência de faixa vermelha. 2 É proibida a utilização de cores nos rótulos que possam causar confusão ou erro na identificação da faixa preta. 315
316 Art. 33. Na faixa vermelha devem ser utilizadas as especificações definidas nesta Resolução para os medicamentos com venda sob prescrição médica. Art. 34. No interior da faixa dos medicamentos à base de substâncias sujeitas a controle especial, devem ser incluídas, em caixa alta, as frases definidas em norma específica e suas atualizações, para a substância ou lista à qual pertence. Art. 35. Os rótulos das embalagens primárias dos medicamentos a base de substâncias sujeitas a controle especial devem possuir as frases definidas em norma específica e suas atualizações, para a substância ou lista a qual pertence. Seção IV - Dos medicamentos com destinação hospitalar Art. 36. Os rótulos das embalagens secundárias dos medicamentos com destinação exclusivamente hospitalar devem possuir a frase, em caixa alta, "EMBALAGEM HOSPITALAR", com tamanho mínimo de 30% da altura do maior caractere do nome comercial ou, na sua ausência, da denominação genérica. Seção V - Dos medicamentos de uso restrito a hospitais Art. 37. Os rótulos das embalagens secundárias de todos os medicamentos com uso restrito a hospitais, definidos em norma específica,devem possuir a frase, em caixa alta, "USO RESTRITO A HOSPITAIS", com tamanho mínimo de 30% da altura do maior caractere do nome comercial ou, na sua ausência, da denominação genérica. 1 A frase deve ser disposta logo acima da faixa de restrição de prescrição, na face principal da embalagem. 2 No caso de medicamentos com destinação hospitalar, a frase "USO RESTRITO A HOSPITAIS" dispensa a inclusão da frase "EMBALAGEM HOSPITALAR". Seção VI - Dos medicamentos oriundos dos Laboratórios Oficiais Art. 38. No caso de medicamentos oriundos de Laboratórios Oficiais para os quais são disponibilizados Mementos Terapêuticos ao invés de bulas para os profissionais de saúde, seguindo o estabelecido em norma específica, substituir a frase "Informações ao profissional de saúde, indicações, contra-indicações e precauções: vide bula" exigida para os rótulos das embalagens secundárias, por "Informações ao profissional de saúde, indicações, contraindicações e precauções: vide Memento Terapêutico". Seção VII - Dos medicamentos com destinação institucional Art. 39. Os rótulos das embalagens primárias e secundárias de todos os medicamentos com destinação institucional, independente da restrição de prescrição, devem possuir a frase, em caixa alta, "PROIBIDA VENDA AO COMÉRCIO", com tamanho mínimo de 30% da altura do maior caractere do nome comercial ou, na sua ausência, da denominação genérica. Parágrafo único. Nos rótulos das embalagens secundárias, a frase deve ser disposta logo acima da faixa de restrição de prescrição, ou em posicionamento equivalente no caso de inexistência da mesma, em sua face principal. 316
317 Art. 40. Os medicamentos com destinação institucional e de venda sob prescrição médica, com ou sem retenção de receita, podemsubstituir a palavra "VENDA" por "USO" nas frases exigidas para os rótulos das embalagens primárias e secundárias. Seção VIII- Dos medicamentos destinados ao Ministério da Saúde Art. 41. Os rótulos das embalagens dos medicamentos com destinação institucional destinados ao Ministério da Saúde, para distribuição através de programas de saúde pública, devem obedecer à identificação padronizada e descrita no Manual de Identificação Visual para Embalagens de Medicamentos, instituído por norma específica. Seção IX - Dos medicamentos genéricos Art. 42. Para os medicamentos genéricos, deve ser adotada para sua identificação, a denominação genérica de cada princípioativo, em letras minúsculas, utilizando a Denominação Comum Brasileira (DCB), sendo expressamente proibido o uso de nome comercial. Art. 43. Os rótulos de todas as embalagens dos medicamentos genéricos devem possuir, com tamanho de 30% da altura do maior caractere da denominação genérica, localizada imediatamente abaixo desta e com o mesmo destaque, a frase "Medicamento genérico Lei nº 9.787, de 1999". Art. 44. Os rótulos das embalagens secundárias dos medicamentos genéricos devem possuir o logotipo que os identifica, impresso dentro de uma faixa amarela disposta em sua face principal e faces laterais, no seu terço médio inferior e com largura não inferior a um quinto da maior face. 1 É proibido colocar a faixa no rodapé das embalagens, devendo-se respeitar o limite mínimo de 10 mm nas bases das embalagens ou na extremidade contrária a sua abertura. 2 Nas embalagens secundárias de medicamentos de venda sob prescrição médica, com ou sem retenção de receita, a faixa amarela deve ficar justaposta logo acima da faixa vermelha. 3 Nas embalagens secundárias de medicamentos à base de substâncias sujeitas a controle especial para as quais é exigida a faixa preta, constantes na norma específica e suas atualizações, a faixa amarela deve ficar justaposta logo abaixo da faixa preta. 4 Nas embalagens secundárias de medicamentos que podem ser vendidos sem exigência de prescrição médica, a faixa amarela deve estar no local correspondente ao que seria o da faixa vermelha. Art. 45. Nas embalagens primárias dos medicamentos sem exigência de prescrição médica, que sejam disponibilizados em embalagens múltiplas e comercializados exclusivamente em embalagem primária, deve ser incluída a faixa amarela com o logotipo do medicamento genérico. Art. 46. Na faixa amarela, deve ser utilizada a referência de cor amarela PANTONE 116C, que pode ser obtida através da mistura de pigmentos de qualquer fabricante de tintas, com variações máximas e mínimas aceitáveis para este tom, e ser aplicado um verniz sobre ela. 1 É proibida a utilização da cor amarela PANTONE 116C fora da faixa amarela e em embalagens de medicamentos que não sejam genéricos. 317
318 2 É proibida a utilização de cores nos rótulos que possam causar confusão ou erro na identificação da faixa amarela. Art. 47. O logotipo do medicamento genérico consiste em uma letra "G" estilizada e as palavras "Medicamento Genérico" escritas na cor azul PANTONE 276C, inseridas em um retângulo amarelo PANTONE 116C. 1 As palavras "Medicamento Genérico" devem ser escritas com a letra tipo "Frutiger Bold Condensed". 2 A palavra "Medicamento" deve ter o mesmo comprimento da palavra "Genérico", ou seja, a letra "M" deve iniciar no mesmo ponto da letra "G" e as letras "o" devem terminar nos mesmos pontos. 3 O logotipo pode ser disposto na versão horizontal e deve ser composto pelas palavras "Medicamento" escrito logo acima da palavra "Genérico", precedido pela letra "G", conforme modelo no Anexo I desta Resolução. 4 O logotipo pode ser disposto na versão vertical e deve ser composto pela letra "G", pela palavra "Medicamento", escrita logo abaixo e pela palavra "Genérico" logo abaixo desta, conforme modelo no Anexo I desta Resolução. Art. 48. O tamanho do logotipo de medicamento genérico é variável conforme o tamanho da face principal da embalagem secundária do medicamento, entretanto, todas as proporções estabelecidas no logotipo devem ser rigorosamente mantidas, conforme Anexo I desta Resolução. 1 Para embalagens de orientação horizontal deve ser utilizada a versão vertical do logotipo com as seguintes características: I - a largura (w) deve ser igual a um quinto da largura da maior face; e II - a altura (h) deve ser igual a 1,25 w. 2 Para embalagens de orientação vertical deve ser utilizada a versão horizontal do logotipo, onde o retângulo tem as seguintes dimensões: I - a altura (h) deve ser um quinto da altura da maior face; e II - a largura (w) deve ser igual a 2,5 h. Art. 49. É permitido imprimir informações exigidas para os rótulos nas laterais da faixa amarela, caso necessário. Seção X - Dos medicamentos dinamizados Art. 50. Os rótulos das embalagens dos medicamentos dinamizados devem conter a frase, conforme a categoria do medicamento, em negrito: "Medicamento Homeopático", "Medicamento Antroposófico" ou "Medicamento Antihomotóxico". Art. 51. Os rótulos das embalagens dos medicamentos dinamizados devem atender ao disposto em normas específicas sobre o registro e notificação de medicamentos dinamizados, além do disposto nesta Resolução. Seção XI - Dos medicamentos fitoterápicos 318
319 Art. 52. Os rótulos das embalagens de medicamentos fitoterápicos devem conter a frase "MEDICAMENTO FITOTERÁPICO", em caixa alta e com tamanho mínimo de 30% da altura do maior caractere do nome comercial. Art. 53. Os medicamentos fitoterápicos que utilizarem como princípios ativos derivados vegetais, como extrato, suco e óleo, podem especificá-los logo após ou abaixo do nome botânico. Seção XII - Dos medicamentos para reconstituição e uso oral Art. 54. No caso de medicamentos nas formas farmacêuticas pó ou granulado, para suspensão ou solução, de uso oral, deve-se: I - indicar na embalagem primária a posição precisa, de forma clara e de fácil leitura, até onde o usuário deve acrescentar o diluente; II - inserir a frase "Modo de preparar: vide bula", no rótulo da embalagem secundária e primária; III - inserir a frase: "Após preparo, manter por ", indicando o cuidado de conservação e prazo de validade da solução ou suspensão reconstituída, no rótulo da embalagem primária ou da secundária, no caso de limitação no campo de impressão da embalagem primária, caso o cuidado de conservação do medicamento depois de preparado diferir do cuidado de conservação antes de aberto. Seção XIII - Dos medicamentos com prazo de validade alterado após aberto Art. 55. No caso de medicamentos cujo prazo de validade original reduzir após aberto, inserir a frase: "Após aberto, valido por ", indicando após de validade após aberto, no rótulo da embalagem primária ou da secundária, no caso de limitação no campo de impressão da embalagem primária. Seção XIV - Dos medicamentos para Terapia de Reidratação Oral (TRO) Art. 56. Nos rótulos das embalagens dos medicamentos para Terapia de Reidratação Oral (TRO) deve ser expressa a quantidade dos princípios ativos em unidades de massa ou massa/volume, e na forma de meq/l. 1 Em caso de concentração de sódio entre 40 e 60 meq/l, deve-se incluir a frase "Para prevenção da desidratação e manutenção da hidratação após a fase de reidratação". 2 Quando o teor de sódio for igual a 90 meq/l, deve-se incluir a frase "Para reidratação e manutenção da hidratação". Seção XV - Das Soluções Parenterais de Pequeno Volume (SPPV) Art. 57. Os rótulos das embalagens primárias das SPPV devem apresentar dimensões de modo a envolver, no máximo, 3/4 da área total do corpo do recipiente e o espaço livre para permitir a visualização do conteúdo do recipiente deve ser no sentido longitudinal do mesmo e ocupar a maior área possível, conforme figura 1 do Anexo II. Art. 58. As informações impressas no rótulo da embalagem primária das SPPV devem estar dispostas paralelamente ao maior eixo do recipiente, com a margem esquerda do rótulo começando o mais próximo possível da base, e devem 319
320 permitir a leitura integral do texto quando o recipiente for seguro pela haste ou gargalo, conforme figura 2 do Anexo II. 1 Quando o nome comercial, a denominação genérica do princípio ativo, a concentração e volume total puderem ser impressos dentro de 180º da circunferência do recipiente, a impressão pode ser feita de forma perpendicular ao seu maior eixo, de acordo com a figura 3 do Anexo II. 2 Para seringas preenchidas, o texto deve ser orientado no sentido "agulha - êmbolo" e de forma a não comprometer a visualização da sua graduação. Art. 59. Para a rotulagem das embalagens primárias das SPPV contendo as substâncias definidas em instrução normativa específica, deve ser respeitada a padronização de cores para a gravação dos dizeres estabelecida na norma específica. Art. 60. As ampolas de vidro dos medicamentos, definidos em instrução normativa específica, devem ser identificadas por dois anéis de cor estabelecida na norma específica, impressos na haste, com largura mínima de 0,6 mm. 1 Quando o medicamento for constituído por apenas um princípio ativo, os dois anéis devem ser da mesma cor indicada para a família. 2 Quando se tratar de associação com no máximo dois princípios ativos, cada anel deve corresponder à cor indicada para a respectiva família. 3 No caso do princípio ativo ser um antagonista, um dos anéis deve ser branco e o outro da cor indicada para a família do princípio ativo a ser antagonizado. Art. 61. As embalagens de SPPV que não permitam a identificação por anéis devem ser diferenciados pelos critérios de cores de impressão no rótulo e colocação de faixa com largura mínima de 3mm na parte superior do rótulo, com a cor correspondente a do anel de ruptura, definida em instrução normativa específica. Seção XVI - Das Soluções Parenterais de Grande Volume (SPGV) Art. 62. Os rótulos das embalagens das SPGV, além das informações mínimas exigidas nesta Resolução, devem conter: I - a composição qualitativa e quantitativa, percentual; II - conteúdo eletrolítico em meq/l ou mmol/l; e, III - osmolaridade; e IV - volume total. Art. 63. No caso da SPGV, de sistema fechado, que possuam apenas um sítio destinado a colocação do equipo, devese incluir a frase "Não é indicada a adição de outro medicamento." Seção XVII - Dos Concentrados Polieletrolíticos para Hemodiálise (CPHD) Art. 64. Os rótulos das embalagens dos CPHD devem apresentar: I - faixa vermelha, com a cor vermelha PANTONE VERMELHO 485 C e largura correspondente a um quinto da maior face do rótulo, com os dizeres "Uso sob prescrição médica"; 320
321 II - faixa no cabeçalho, com largura correspondente a um quinto da menor face do rótulo, de cor azul PANTONE BLUE 072 C. Parágrafo único. Deve ser respeitado o limite mínimo de 10 mm nas bases das rotulagens, como caracterização daquilo que se entende como rodapé. Art. 65. Para a denominação genérica dos CPHD, utilizar o nome de dois sais da formulação seguidos da expressão: "+ ASSOCIAÇÃO". Parágrafo único. A seqüência de sais a ser utilizada deve seguir a ordem: sódio, potássio e cálcio. Art. 66. Os rótulos das embalagens dos CPHD, além das informações mínimas exigidas nesta Resolução, devem conter: I - composição qualitativa, de acordo com a Denominação Comum Brasileira (DCB), e quantitativa dos sais expressas em p/v (g/l) ou p/p (g/g) no concentrado e meq/l dos íons ou mmol/l das moléculas, após diluição, atendendo aos limites estabelecidos no Anexo III; II - o modo de preparo, incluindo a proporção de diluição a ser empregada. Art. 67. Nos rótulos das embalagens dos CPHD deve ser incluída, em negrito, a frase "USO RESTRITO EM HEMODIÁLISE". Seção XVIII - Dos polivitamínicos, poliminerais e poliaminoácidos Art. 68. Nos rótulos das embalagens dos polivitamínicos, poliminerais e poliaminoácidos, deve constar a formulação qualitativa e quantitativa por unidade farmacotécnica e o teor percentual de cada princípio ativo na dose/posologia diária máxima preconizada, expresso claramente em índices percentuais, relativos à Ingestão Diária Recomendada (IDR). Seção XIX - Dos medicamentos com envoltórios intermediários Art. 69. Os envoltórios intermediários devem possuir todas as informações mínimas exigidas para as embalagens primárias, quando ele impedir a visualização das informações dispostas nas embalagens primárias. Parágrafo único. Quando o envoltório intermediário é utilizado para garantir a estabilidade do medicamento, conforme demonstrado em estudo de estabilidade, ele deve possuir a frase: "Apenas remover o envoltório para o uso". Seção XX - Dos medicamentos com duas ou mais apresentações para uso concomitante ou seqüencial Art. 70. As embalagens secundárias e primárias dos medicamentos com duas ou mais apresentações para uso concomitante ou seqüencial devem conter as suas datas de fabricação, validade e número de lote. 1 O numero do lote da apresentação final deve ser correspondente ao da montagem do conjunto das apresentações para uso concomitante ou seqüencial. 321
322 2 A data de fabricação do conjunto das apresentações deve ser a data da montagem do conjunto das apresentações para uso concomitante ou seqüencial. 3 A data de validade da apresentação final deve ser a data da primeira apresentação a vencer. Art. 71. Os rótulos das embalagens primárias das formas farmacêuticas sólidas ou outras previamente fracionadas para uso concomitante ou seqüencial devem possuir na parte frontal um retângulo, ou outra marcação divisória, em destaque, incluindo e indicando as unidades farmacotécnicas a serem administradas por dose e, no verso, devem constar a denominação genérica de cada princípio ativo e concentrações correspondentes àquela unidade farmacotécnica. Seção XXI - Dos medicamentos com dessecantes desprendidos em sua embalagem Art. 72. Quando na embalagem do medicamento houver dessecantes desprendidos em seu interior, na própria unidade do dessecante devem constar: I - os dizeres, em caixa alta: "PERIGO. NÃO COMER"; II - a frase "Conteúdo:.", indicando a substância que constitui o dessecante. Art. 73. É facultativo usar imagem, com no mínimo 10 mm de diâmetro, de um rosto de boca aberta ingerindo um sólido dentro de um círculo com uma faixa diagonal, ambos na cor vermelha, ou, de uma caveira, com ossos cruzados atrás ou abaixo do crânio da caveira, ambos de cor preta, conforme figuras do Anexo IV, informando sobre a proibição de ingestão do dessecante, CAPÍTULO IV - AS ALTERAÇÕES DE ROTULAGEM DE MEDICAMENTOS Seção I - Das notificações de alterações de rotulagem Art. 74. São passíveis de notificação de alteração de rotulagem, com implementação imediata sem manifestação prévia da Anvisa, as atualizações de informações nos rótulos a seguir relacionadas: I - à Lista de Denominação Comum Brasileira (DCB); II - ao Vocabulário Controlado; III - ao novo enquadramento dos medicamentos quanto à restrição de uso e prescrição que venha a ser exigida em norma específica; IV - à incorporação de frases de alerta que venha a ser exigida em norma específica; V - aos Dizeres Legais, quanto ao Telefone do Serviço de Atendimento ao Consumidor (SAC), e ao nome do responsável técnico, número de inscrição e sigla do Conselho Regional de Farmácia; VI - aos Dizeres Legais, quanto à razão social das empresas internacionais; e VII - aos Dizeres Legais, quanto à razão social das empresas nacionais, após aprovação da Anvisa da alteração de razão social. Art. 75. Após qualquer alteração de rotulagem, relacionada à notificação de alteração de rotulagem, as empresas terão um prazo máximo de 180 (cento e oitenta) dias para começarem a fabricar medicamentos com os novos rótulos a partir da data da notificação, sendo este o tempo previsto para esgotamento de estoque. Seção II - Das alterações de rotulagem relacionadas ao pósregistro e renovação 322
323 Art. 76. Após qualquer alteração de pós-registro e renovação que envolva adequação de informação na rotulagem, as empresas terão um prazo máximo de 180 (cento e oitenta) dias para começarem a fabricar medicamentos com os novos rótulos, a partir da aprovação da petição pela Anvisa, sendo este o tempo previsto para esgotamento de estoque. Art. 77. No caso de alterações qualitativas excipientes aprovadas pela Anvisa, em que a empresa desejar manter o nome comercial e a mesma indicação terapêutica, os rótulos das embalagens secundárias e, na sua ausência, da primária, devem conter a frase "Nova Fórmula" próxima ao nome comercial ou, na sua ausência, da denominação genérica, com tamanho mínimo de 30% da altura do seu maior caractere. Parágrafo único. Esta frase é obrigatória por no mínimo um ano depois de aprovada a alteração. Seção III - Da inclusão de informações não previstas nos rótulos de medicamentos Art. 78. Caso seja identificada a necessidade de incluir alguma informação não prevista nesta Resolução nos rótulos de medicamentos, as empresas deverão peticionar uma alteração de rotulagem acompanhada de justificativa técnica. 1 A inclusão de informações somente poderá ser implementada com a aprovação expressa da Anvisa, que será publicada em Diário Oficial da União (DOU). 2 As empresas terão um prazo máximo de 180 (cento e oitenta) dias para começarem a fabricar medicamentos com os novos rótulos a partir da aprovação da petição pela Anvisa, sendo este o tempo previsto para esgotamento de estoque. Seção IV - Da inclusão de rótulo para apresentações com nova destinação Art. 79. Para incluir novo rótulo para apresentação de medicamento anteriormente registrada que contemplará nova destinação, deve ser peticionada a inclusão de rotulagem. Parágrafo único. O novo rótulo poderá diferir do rótulo aprovado anteriormente apenas no que se refere às informações específicas exigidas para a nova destinação. CAPÍTULO V - DAS DISPOSIÇÕES FINAIS E TRANSITORIAS Art. 80. As empresas devem notificar a adequação da rotulagem, atendendo ao disposto nesta Resolução, e disponibilizar os novos rótulos nas embalagens dos medicamentos fabricados ou importados para venda no mercado nacional em até 540 (quinhentos e quarenta) dias a partir da publicação desta Resolução. (Prazo suspenso pela RESOLUÇÃO - RDC Nº 26, DE 16 DE JUNHO DE 2011) 1 Os novos rótulos deverão contemplar informações em conformidade com as aprovadas no registro, pós-registro ou renovação dos medicamentos. 2 Os novos rótulos deverão ser disponibilizados no prazo previsto no caput deste artigo independentemente de manifestação prévia da Anvisa. 3 As notificações de adequação das rotulagens serão verificadas durante a análise de pós-registro e renovações de registro, momento no qual poderão ser feitas exigências caso a rotulagem não se enquadre no estabelecido nesta Resolução. 323
324 Art. 81. Compete à autoridade sanitária estadual, municipal e federal proceder, nas inspeções rotineiras nas indústrias farmacêuticas ou importadoras de medicamentos, à verificação dos rótulos dos medicamentos, conforme o disposto nesta Resolução e aprovado no registro, pós-registro e renovação dos medicamentos, respeitando-se o prazo de adequação. Art. 82. O descumprimento das disposições contidas nesta Resolução e no regulamento por ela aprovado constituem infração sanitária, nos termos da Lei n 6.437, de 20 de agosto de 1977, sem prejuízo das responsabilidades civil, administrativa e penal cabíveis. Art. 83. Ficam revogados os artigos 4 e 5 da Portaria nº 802, de 8 de outubro de 1998; a Resolução - RDC n 9, de 2 de janeiro de 2001; e a Resolução - RDC nº 333, de 19 de novembro de 2003, com exceção do item 3 do Anexo, referente à regulamentação de nomes comerciais, e do item 17 do Anexo, cuja vigência foi restabelecida pela Resolução - RDC nº 60 de 26 de novembro de 2009 durante o período de transição de 90 dias. Art. 84. Esta Resolução entra em vigor na data de sua publicação. 324
325 ANEXO I - MANUAL DE IDENTIDADE VISUAL PARA A ROTULAGEM DE MEDICAMENTOS GENÉRICOS 325
326 326
327 327
328 328
329 329

330 330
331 331
332 332
333 ANEXO II FIGURA 1 - AMPOLA COM REPRESENTAÇÃO DO RÓTULO. 333
334 FIGURA 2 - IMPRESSO PARALELO E PERPENDICULAR AO MAIOR EIXO DO RECIPIENTE FIGURA 3 - IMPRESSO PARALELO E PERPENDICULAR AO MAIOR EIXO DO RECIPIENTE 334
335 ANEXO III TABELA 1 - SOLUÇÕES CONCENTRADAS COM ACETATO OU LACTATO MMol / L meq/l Acetato ou lactato (equivalente a bicarbonato) 32,0-45,0 32,0-45,0 Cálcio 1,0-2,0 2,0-4,0 Cloreto 90, ,0-120 Magnésio 0,25-1,2 0,5-2,4 Potássio 0,0-3,0 0,0-3,0 Sódio 130,0-145,0 Glicose 0,0-12,0 130,0-145,0 TABELA 2 - SOLUÇÕES CONCENTRADAS ÁCIDAS, SEM ADIÇÃO DE BICARBONATO: MMol / L meq/l Ácido Acético 2,5-10,0 2,5-10,0 Cálcio 1,0-2,0 2,0-4,0 Cloreto 90, ,0-120 Magnésio 0,25-1,2 0,5-2,4 Potássio 0,0-3,0 0,0-3,0 Sódio 80,0-110,0 Glicose 0,0-12,0 80,0-110,0 NOTA: Soluções de bicarbonato de sódio ou bicarbonato de sódio sólido, para serem adicionadas imediatamente antes do uso, devem ser fornecidas em quantidades que não ultrapassem a concentração de 45 mmol/l ou 45 meq/l no produto diluído. Neste caso, deve considerar o somatório dos íons sódio na concentração final diluída das soluções concentradas ácidas. 335
336 ANEXO IV FIGURA 1 - IMAGENS PARA DESSECANTES 336
337 RDC Nº 59, DE 10 DE OUTUBRO DE 2014 Dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das atribuições que lhe confere os incisos III e IV, do art. 15 da Lei n.º 9.782, de 26 de janeiro de 1999, o inciso V, e 1 e 3 do art. 5 do Regimento Interno aprovado nos termos do Anexo I da Portaria nº 650 da ANVISA, de 29 de maio de 2014, tendo em vista os incisos III, do art. 2º, III e IV, do art. 7º da Lei nº 9.782, de 1999, o Programa de Melhoria do Processo de Regulamentação da Agência, instituído por meio da Portaria nº 422, de 16 de abril de 2008, em reunião realizada em 07 de outubro de 2014, adota a seguinte Resolução da Diretoria Colegiada e eu, Diretor- Presidente, determino a sua publicação: Art. 1º O nome dos medicamentos e seus complementos, deverão atender aos critérios desta resolução. Capítulo I - Das disposições gerais Art. 2º Esta Resolução apresenta os critérios para a formação dos nomes dos medicamentos. Art. 3º Este Regulamento se aplica a todos os medicamentos sujeitos ao registro ou notificação simplificada, excetuando-se os medicamentos genéricos e imunoterápicos. Art. 4º Para efeito deste regulamento técnico são adotadas as seguintes definições: I - nome de medicamento: é a designação do produto farmacêutico tecnicamente elaborado, para distingui-lo de outros; II - fármaco(s) identificador(es): é o insumo farmacêutico ativo, ou conjunto destes em uma associação, responsável pela indicação terapêutica principal, presente em todos os integrantes de uma determinada família de medicamentos; III - complemento de nome: palavra empregada como designação complementar, de uso não exclusivo, ao nome do medicamento; e IV - família de medicamentos: conjunto de produtos farmacêuticos de uma mesma empresa, com mesmo(s) fármaco(s) identificador( es), agrupados por um nome comum e diferenciados por complementos individuais. Capítulo II - Das famílias de medicamentos Art. 5º Os medicamentos da mesma empresa, cuja formulação contenha pelo menos um fármaco identificador, poderão ser agrupados em famílias compartilhando um nome comum e adotando complementos diferenciadores que os distingam. 1º A exclusão ou a substituição de um ou mais fármacos identificadores enseja a adoção de um nome distinto para tal medicamento. 2º Para produtos farmacêuticos polivitamínicos, poliminerais e/ou poliaminoácidos é admitida a mudança de parte da composição, mantida a indicação terapêutica original, visando à adequação do medicamento a populações-alvo específicas. Art. 6º Na hipótese de formação de famílias, a empresa deverá adotar medidas distintivas complementares por meio da rotulagem dos produtos envolvidos, de forma a promover maior distinção entre eles. 337
338 Capítulo III - Dos nomes dos medicamentos Seção I - Critérios para nomes de medicamentos Art. 7º O nome de medicamento deve, preferencialmente, ser composto por uma única palavra e sua pronúncia pretendida no idioma português deve guardar relação direta com sua grafia. Parágrafo único. O nome pretendido deve guardar suficiente distinção gráfica e fonética em relação às designações de outros medicamentos já registrados. Art.8º O nome de medicamentos isentos de prescrição médica poderá evocar a indicação terapêutica principal aprovada. Art. 9º As vitaminas, minerais ou aminoácidos, isolados ou associados entre si, poderão adotar, para seus componentes, os sinônimos usuais utilizados na literatura técnica, desde que não exista controvérsia quanto a sua identidade. Art. 10. Os fitoterápicos poderão adotar o nome popular, científico ou sinônimo usual na literatura técnica como nome do medicamento, desde que não exista controvérsia quanto a sua identidade, acrescido do nome da empresa e seguido da nomenclatura botânica que inclui gênero e espécie. Art. 11. Os laboratórios oficiais mantidos pelo poder público podem adotar o próprio nome ou sigla característica junto ao nome do medicamento ou designação genérica. Art. 12. É facultada a utilização da denominação genérica, conforme a Denominação Comum Brasileira (DCB) ou, em sua ausência, a Denominação Comum Internacional (DCI), para designar as soluções parenterais de pequeno volume (SPPV) e soluções parenterais de grande volume (SPGV) unitárias isentas de fármacos, registradas como medicamento específico. Art. 13. O Concentrado Polieletrolítico para Hemodiálise poderá adotar esta designação ou a abreviatura CPHD, precedida do nome da empresa. Art. 14. Os medicamentos dinamizados adotarão, além do disposto nesta resolução, as disposições específicas contidas nas respectivas farmacopeias ou compêndios aceitos pela ANVISA. Art. 15. Os nomes dos medicamentos e seus complementos não poderão empregar: I - os sufixos da denominação comum recomendada para cada classe terapêutica de substâncias farmacêuticas, ainda que em posição distinta da usualmente recomendada, dentro da própria classe química ou não; II - a parte da denominação comum do fármaco, não referida no inciso I, usualmente associada a determinado princípio ativo, quando este não fizer parte da composição do medicamento; III - abreviaturas, letras isoladas, sequências aleatórias de letras, algarismos arábicos ou romanos, inclusive por extenso, sem significado evidente ao consumidor ou não possuam relação com as características do produto, no caso dos complementos diferenciais; IV - designações que não correspondam à forma farmacêutica do medicamento em questão; 338
339 V - palavras ou expressões que possam induzir ao entendimento de que o medicamento seja inócuo, natural, isento ou com reduzidos efeitos colaterais, ou possua potência e qualidade superiores, propriedades especiais não comprovadas; ou VI - palavras ou expressões que valorizem uma ação terapêutica, sem comprovação mediante estudos clínicos, e possam induzir o consumidor a entender que tal medicamento teria efeito terapêutico superior a outro medicamento de igual composição; VII - nome de medicamento que foi indeferido por motivação de eficácia e segurança, exceto quando de mesma indicação terapêutica. Parágrafo único. A ANVISA na avaliação de outras hipóteses não previstas neste artigo poderá, mediante motivação de risco ao consumidor, recusar o nome do medicamento pleiteado. Seção II -Critérios para complementos de nomes de medicamentos. Art. 16. Os complementos de nome devem ser utilizados com o objetivo de distinguir um determinado produto farmacêutico de outro já registrado pela mesma empresa, dentro de uma família de medicamentos. 1º A Anvisa não considerará, para fins de registro, a exclusividade de utilização de complementos de nome. 2º É vedada a utilização do mesmo complemento de nome com significados distintos. 3º Poderão ser utilizados complementos de nome para distinção de vias de administração, forma farmacêutica, populaçãoalvo, absorção ou ainda outras situações, mediante justificativa fundamentada da empresa. 4º Deverão adotar complementos de nome aqueles medicamentos que apresentem cinética de liberação distinta, forma farmacêutica distinta ou via de administração distinta dentro de uma mesma família de medicamentos. Art. 17. A empresa interessada deverá, ao propor a utilização de um determinado complemento de nome, justificar tecnicamente seu pleito, destacando como ele auxilia na distinção entre os medicamentos de uma mesma família ou outros medicamentos de denominação assemelhada. Capítulo IV - Das disposições finais e transitórias Art. 18. É responsabilidade da empresa proceder à avaliação do nome pretendido para dado medicamento diante dos critérios apresentados nesta Resolução, bem como à comparação com outros medicamentos da mesma categoria terapêutica ou outras categorias afinas antes da submissão da proposta, buscando garantir a não colidência do nome proposto com outros já registrados. Art. 19. É responsabilidade do titular do registro sanitário dar ampla publicidade a qualquer alteração de nome do medicamento ou seu complemento, decorrente do disposto nesta resolução. Art. 20. Os nomes de medicamentos aprovados mediante publicação do respectivo registro sanitário pela ANVISA, que estejam de acordo com a regulamentação anterior à vigência desta Resolução, não serão objeto de revisão por parte da agência. 339
340 Parágrafo único. Constatado potencial risco sanitário relacionado ao nome de um ou mais medicamentos, seja por vício na concessão do mesmo ou por fatos supervenientes que possam indicar a possibilidade de que este induza a erro, será instruído processo administrativo para fundamentação e avaliação do mesmo. Art. 21. É facultado ao titular de registro sanitário, a qualquer tempo, solicitar a alteração de nome de medicamento de sua propriedade, com vistas à adequação aos termos deste regulamento. Art. 22. Os casos omissos nesta norma serão avaliados pela área técnica competente, ressalvado o direito de recurso do interessado à Diretoria Colegiada da Anvisa. Art. 23. Revoga-se o item 3 da Resolução RDC n.º 333, de 19 de novembro de Art. 24. Esta Resolução entra em vigor na data de sua publicação. RDC Nº 96, DE 17 DE DEZEMBRO DE 2008 Dispõe sobre a propaganda, publicidade, informação e outras práticas cujo objetivo seja a divulgação ou promoção comercial de medicamentos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o artigo 11, inciso IV, do Regulamento da Agência Nacional de Vigilância Sanitária aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, e conforme artigo 11, inciso IV, do Regimento Interno, aprovado nos termos do Anexo I da Portaria nº 354 da Anvisa, de 11 de agosto de 2006, republicada no DOU de 21 de agosto de 2006, em reunião realizada em 21 de novembro de 2008; considerando a Constituição Federal de 1988; considerando a Lei nº 6.360, de 23 de setembro de 1976; considerando o Decreto nº , de 5 de janeiro de 1977, que regulamenta a Lei nº 6.360, de 24 de setembro de 1976; considerando a Lei nº 9.782, de 26, de janeiro de 1999; considerando a Lei nº 9.787, de 10 de fevereiro de 1999; considerando a Lei nº de 23 de agosto de 2006; considerando o Decreto nº , de 21 de dezembro de 1976, que regulamenta a Lei nº 6.368, de 21 de outubro de 1976; considerando a Lei nº 6.437, de 20 de agosto de 1977, sobre infrações sanitárias, considerando a Lei nº de 15 de julho de 1996; considerando o Decreto nº 2.018, de 1º de outubro de 1996, que regulamenta a Lei nº 9.294, de 15 de julho de 1996; considerando a Lei nº 8.078, de 11 de setembro de 1990; considerando o Decreto nº 2.181, de 20 de março de 1997; considerando a Lei nº 8.069, de 13 de julho de 1990; considerando a Lei nº , de 6 de outubro de 2003; considerando a RDC nº 26, de 30 de março de 2007; 340
341 considerando a Portaria nº 3.916, de 30 de outubro de 1998, que define a Política Nacional de Medicamentos; considerando a publicação do Ministério da Saúde e da Agência Nacional de Vigilância Sanitária intitulada Estudo Comparado - Regulamentação da Propaganda de Medicamentos; considerando a necessidade de atualização do Regulamento Técnico sobre Propaganda, Publicidade, Promoção e Informação de Medicamentos; adota a seguinte Resolução de Diretoria Colegiada e eu Diretor-Presidente, determino a sua publicação. Art. 1º O Regulamento anexo a esta Resolução se aplica à propaganda, publicidade, informação e outras práticas cujo objetivo seja a divulgação ou promoção comercial de medicamentos, de produção nacional ou estrangeira, quaisquer que sejam as formas e meios de sua veiculação, incluindo as transmitidas no decorrer da programação normal das emissoras de rádio e televisão. Art. 2º Esta Resolução de Diretoria Colegiada entra em vigor 180 (cento e oitenta) dias após a sua publicação. ANEXO - REGULAMENTO Art. 1º Este Regulamento se aplica à propaganda, publicidade, informação e outras práticas cujo objetivo seja a divulgação ou promoção comercial de medicamentos de produção nacional ou estrangeira, quaisquer que sejam as formas e meios de sua veiculação, incluindo as transmitidas no decorrer da programação normal das emissoras de rádio e televisão. TÍTULO I - REQUISITOS GERAIS Art. 2º Para efeito deste Regulamento são adotadas as seguintes definições: DENOMINAÇÃO COMUM BRASILEIRA/DCB - Denominação do fármaco ou princípio farmacologicamente ativo aprovada pelo órgão federal responsável pela vigilância sanitária. DENOMINAÇÃO COMUM INTERNACIONAL/DCI - Denominação do fármaco ou princípio farmacologicamente ativo recomendada pela Organização Mundial da Saúde. EMPRESA - Pessoa jurídica, de direito público ou privado, que exerça como atividade principal ou subsidiária a produção, manipulação, comércio, fornecimento, distribuição e divulgação de medicamentos, insumos farmacêuticos e outros produtos que sejam anunciados como medicamento. MARCA NOMINATIVA - É aquela constituída por uma ou mais palavras no sentido amplo do alfabeto romano, compreendendo, também, os neologismos e as combinações de letras e/ou algarismos romanos e/ou arábicos. MARCA FIGURATIVA - É aquela constituída por desenho, figura ou qualquer forma estilizada de letra e número, isoladamente. 341
342 MARCA MISTA - É aquela constituída pela combinação de elementos nominativos e figurativos ou de elementos nominativos com grafia apresentada de forma estilizada. MATERIAL CIENTÍFICO - Artigos científicos publicados e livros técnicos. MATERIAL DE AJUDA VISUAL - peça publicitária utilizada exclusivamente pelos propagandistas com o objetivo de apresentar aos profissionais prescritores e dispensadores os medicamentos com informações e linguagem uniformizadas pela empresa. MEDICAMENTO BIOLÓGICO - Medicamento biológico que contém molécula com atividade biológica conhecida, e que tenha passado por todas as etapas de fabricação (formulação, envase, liofilização, rotulagem, embalagem, armazenamento, controle de qualidade e liberação do lote de produto biológico para uso). MENSAGEM RETIFICADORA - É aquela elaborada para esclarecer e corrigir erros e equívocos causados pela veiculação de propagandas enganosas e/ou abusivas, e/ou que apresentem informações incorretas e incompletas sendo, portanto, capazes de induzir, direta ou indiretamente, o consumidor a erro e a se comportar de forma prejudicial à sua saúde e segurança. MONOGRAFIA - Material elaborado mediante uma compilação de informações técnicºcientíficas provenientes de estudos publicados, livros técnicos e informações contidas na documentação de registro submetida à Anvisa, visando munir o profissional de saúde com variadas informações sobre determinado medicamento, apresentando resumos com informações equilibradas, ou seja, resultados satisfatórios e não satisfatórios, e conclusões fiéis à original. NIVEL DE EVIDÊNCIA I - Nível de estudo I: Ensaios clínicos randomizados, com desfecho e magnitude de efeitos clinicamente relevantes, correspondentes à hipótese principal em tese, com adequado poder e mínima possibilidade de erro alfa. Metaanálises de ensaios clínicos de nível II, comparáveis e com validade interna, com adequado poder final e mínima possibilidade de erro alfa. NIVEL DE EVIDÊNCIA II - Nível de estudo II: Ensaio clínico randomizado que não preenche os critérios do nível I. Análise de hipóteses secundárias de estudos nível I. PATROCÍNIO - Custeio total ou parcial da produção de material, programa de rádio ou televisão, evento, projeto comunitário, atividade cultural, artística, esportiva, de pesquisa ou de atualização científica, concedido como estratégia de marketing, bem como custeio dos participantes das atividades citadas. PEÇA PUBLICITÁRIA - Cada um dos elementos produzidos para uma campanha publicitária ou de promoção de vendas, com funções e características próprias, que seguem a especificidade e a linguagem de cada veículo. Exemplos: anúncio, encarte, filmete, spot, jingle, cartaz, cartazete, painel, letreiro, display, folder, banne r, móbile, outdoor, busdoor, visual aid etc. PESSOA FÍSICA - aquela que, de forma direta ou indireta, seja responsável por atividades relacionadas à produção, manipulação, comércio, fornecimento, distribuição e divulgação de medicamentos, insumos farmacêuticos e outros produtos que sejam anunciados como medicamento. PREPARAÇÃO MAGISTRAL - É aquela preparada na farmácia, de forma individualizada, para ser dispensada atendendo a uma prescrição de um profissional habilitado, respeitada a legislação vigente, que estabelece sua composição, forma farmacêutica, posologia e modo de usar. 342
343 PREPARAÇÃO OFICINAL - É aquela preparada na farmácia, cuja fórmula esteja inscrita nas farmacopéias, compêndios ou formulários reconhecidos pelo Ministério da Saúde. PROGRAMAS DE FIDELIZAÇÃO - São aqueles realizados por farmácias e drogarias, as quais, na intenção de fidelizar o consumidor, possibilitam aos clientes, em troca da compra de produtos, a participação em sorteios, ganho de prêmios ou descontos na compra de produtos, entre outros benefícios. PROPAGANDA/PUBLICIDADE - Conjunto de técnicas e atividades de informação e persuasão com o objetivo de divulgar conhecimentos, tornar mais conhecido e/ou prestigiado determinado produto ou marca, visando exercer influência sobre o público por meio de ações que objetivem promover e/ou induzir à prescrição, dispensação, aquisição e utilização de medicamento. PROPAGANDA/PUBLICIDADE ABUSIVA - É aquela que incita a discriminação de qualquer natureza, a violência, explora o medo ou superstições, se aproveita da deficiência de julgamento e de experiência da criança, desrespeita valores ambientais ou que seja capaz de induzir o usuário a se comportar de forma prejudicial ou perigosa à sua saúde ou segurança. PROPAGANDA/PUBLICIDADE ENGANOSA - É qualquer modalidade de informação ou comunicação de caráter publicitário, inteira ou parcialmente falsa, ou que, por qualquer outro modo, mesmo por omissão de dado essencial do produto, seja capaz de induzir o consumidor a erro, a respeito da natureza, características, qualidade, quantidade, propriedades, origem, preço e quaisquer outros dados sobre produtos e serviços. PROPAGANDA/PUBLICIDADE INDIRETA - É aquela que, sem mencionar o nome dos produtos, utiliza marcas, símbolos, designações e/ou indicações capaz de identificá-los e/ou que cita a existência de algum tipo de tratamento para uma condição específica de saúde. REFERÊNCIA BIBLIOGRÁFICA - Conjunto padronizado de elementos descritivos que permite a identificação de documentos utilizados, possibilitando sua localização e obtenção direta por um leitor interessado. SUBSTÂNCIA ATIVA - Qualquer substância que apresente atividade farmacológica ou outro efeito direto no diagnóstico, tais como: cura, alívio, tratamento ou prevenção de doenças; ou afete qualquer função do organismo humano. VACINAS - Produtos biológicos que contêm uma ou mais substâncias antigênicas que, quando inoculados, são capazes de induzir imunidade específica ativa e proteger contra a doença causada pelo agente infeccioso que originou o antígeno. Art. 3º Somente é permitida a propaganda ou publicidade de medicamentos regularizados na Anvisa. 1º A propaganda ou publicidade deve ser procedente de empresas regularizadas perante o órgão sanitário competente, quando assim a legislação o exigir, ainda que a peça publicitária steja de acordo com este Regulamento. 2º Todas as alegações presentes na peça publicitária referentes à ação do medicamento, indicações, posologia, modo de usar, reações adversas, eficácia, segurança, qualidade e demais características do medicamento devem ser compatíveis com as informações registradas na Anvisa. 343
344 3º As referências bibliográficas, citadas na propaganda ou publicidade de medicamentos, devem estar disponíveis no Serviço de Atendimento ao Consumidor (SAC) e aos profissionais prescritores e dispensadores de medicamentos. 3º - O conteúdo das referências bibliográficas citadas na propaganda ou publicidade de medicamentos isentos de prescrição devem estar disponíveis pela empresa no Serviço de Atendimento ao Consumidor (SAC) e no serviço de atendimento aos profissionais prescritores e dispensadores de medicamentos. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) 4º - O conteúdo das referências bibliográficas citadas na propaganda ou publicidade de medicamentos de venda sob prescrição devem estar disponíveis no serviço de atendimento aos profissionais prescritores e dispensadores de medicamentos. (Parágrafo incluído pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 4º Não é permitida a propaganda ou publicidade enganosa, abusiva e/ou indireta. Parágrafo único - Fica vedado utilizar técnicas de comunicação que permitam a veiculação de imagem e/ou menção de qualquer substância ativa ou marca de medicamentos, de forma não declaradamente publicitária, de maneira direta ou indireta, em espaços editoriais na televisão; contexto cênico de telenovelas; espetáculos teatrais; filmes; mensagens ou programas radiofônicos; entre outros tipos de mídia eletrônica ou impressa. Art. 5º As empresas não podem outorgar, oferecer, prometer ou distribuir brindes, benefícios e vantagens aos profissionais prescritores ou dispensadores, aos que exerçam atividade de venda direta ao consumidor, bem como ao público em geral. (Vide a INSTRUÇÃO NORMATIVA Nº 05, DE 20 DE MAIO DE 2009) Art. 6º As informações exigidas neste Regulamento, quando exibidas em linguagem escrita, devem ser apresentadas em cores que contrastem com o fundo do anúncio, devem estar dispostas no sentido predominante da leitura da peça publicitária e devem permitir a sua imediata visualização, guardando entre si as devidas proporções de distância, indispensáveis à legibilidade e destaque. Parágrafo único: No caso de propaganda ou publicidade veiculada na televisão, quando as informações escritas não forem locucionadas, elas deverão ser exibidas pelo tempo suficiente à leitura. Art. 7º As informações sobre medicamentos devem ser comprovadas cientificamente. Art. 8º É vedado na propaganda ou publicidade de medicamentos: I - estimular e/ou induzir o uso indiscriminado de medicamentos; II - sugerir ou estimular diagnósticos ao público em geral; III - incluir imagens de pessoas fazendo uso do medicamento; IV - anunciar um medicamento como novo, depois de transcorridos dois anos da data de início de sua comercialização no Brasil; 344
345 V - incluir selos, marcas nominativas, figurativas ou mistas de instituições governamentais, entidades filantrópicas, fundações, associações e/ou sociedades médicas, organizações não-governamentais, associações que representem os interesses dos consumidores ou dos profissionais de saúde e/ou selos de certificação de qualidade; VI - sugerir que o medicamento possua características organolépticas agradáveis, tais como: "saboroso", "gostoso", "delicioso" ou expressões equivalentes; bem como a inclusão de imagens ou figuras que remetam à indicação do sabor do medicamento; VII - empregar imperativos que induzam diretamente ao consumo de medicamentos, tais como: "tenha", "tome", "use", "experimente"; VIII - fazer propaganda ou publicidade de medicamentos e (ou) empresas em qualquer parte do bloco de receituários médicos; IX - criar expectativa de venda; X - divulgar como genéricos os medicamentos manipulados ou industrializados que não sejam genéricos, nos termos da Lei nº 9.787/99; XI - usar expressões ou imagens que possam sugerir que a saúde de uma pessoa poderá ser afetada por não usar o medicamento. Art. 9º É permitido na propaganda ou publicidade de medicamentos: I - utilizar figuras anatômicas, a fim de orientar o profissional de saúde ou o paciente sobre a correta utilização do produto; I - informar o sabor do medicamento; III - utilizar expressões tais como: "seguro", "eficaz" e "qualidade", em combinação ou isoladamente, desde que complementadas por frases que justifiquem a veracidade da informação, as quais devem ser extraídas de estudos veiculados em publicações científicas e devem estar devidamente referenciadas; IV - utilizar expressões tais como: "absoluta", "excelente", "máxima", "ótima", "perfeita", "total" relacionadas à eficácia e à segurança do medicamento, quando fielmente reproduzidas de estudos veiculados em publicações científicas e devidamente referenciadas; V - quando constar das propriedades aprovadas no registro do medicamento na Anvisa, informar que o medicamento pode ser utilizado por qualquer pessoa, em qualquer faixa etária, inclusive por intermédio de imagens; V - quando constar das propriedades aprovadas no registro do medicamento na Anvisa, informar que o medicamento pode ser utilizado por qualquer faixa etária, inclusive por intermédio de imagens; (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) VI - quando determinado pela Anvisa, publicar mensagens tais como: "Aprovado", "Recomendado por especialista", "o mais freqüentemente recomendado" ou "Publicidade Aprovada pela Vigilância Sanitária'', pelo ''Ministério da Saúde", ou mensagem similar referente a órgão congênere Estadual, Municipal e do Distrito Federal; VII - fazer menção à quantidade de países onde o medicamento é comercializado e/ou fabricado, desde que os países sejam identificados na peça publicitária. Art. 10 Os programas de fidelização realizados em farmácias e drogarias, dirigidos ao consumidor, não podem ter medicamentos como objeto de pontuação, troca, sorteios ou prêmios. 345
346 Parágrafo único - Todo o material publicitário de divulgação e o regulamento dos programas de fidelização devem informar sobre a restrição prevista no caput deste artigo. Art. 11 A comparação de preços dirigida aos consumidores somente poder feita entre medicamentos que sejam intercambiáveis nos termos da Lei nº 9.787/99. 1º Somente aos profissionais prescritores é permitida a comparação de preço entre medicamentos que não sejam intercambiáveis, com base em informações mercadológicas, desde que tenham o mesmo princípio ativo. 2º A comparação deve ser feita entre os custos de tratamento ou, no caso de medicamentos de uso contínuo, entre as doses diárias definidas. 3º A propaganda ou publicidade de medicamentos biológicos, assim classificados conforme regulamento específico, não pode apresentar comparação de preços, mesmo que elas tenham a mesma indicação. 4º Quando informado um valor porcentual do desconto e/ou o preço promocional do medicamento, o preço integral praticado pela farmácia ou drogaria também deve ser informado. 5º Quando as farmácias e drogarias anunciarem descontos para medicamentos, seja por intermédio de anúncios veiculados na televisão, rádio, impressos, faixas ou qualquer outro meio, devem ter disponível, em local visível ao público, lista dos medicamentos anunciados com o preço reduzido conforme artigo 18 deste Regulamento. Art. 12 É permitido oferecer, aos prescritores e dispensadores, material com a relação de medicamentos genéricos em lista que contemple o número de registro na Anvisa, o nome do detentor do registro, a apresentação, incluindo a concentração, a forma farmacêutica e a quantidade, o nome do medicamento de referência e o respectivo detentor do registro, ficando dispensadas as informações dos artigos 22, 23 e 27 deste Regulamento. Art. 12 É permitido oferecer, aos prescritores e dispensadores, material com a relação de medicamentos genéricos em lista que contemple o número de registro na Anvisa, o nome do detentor do registro, a apresentação, incluindo a concentração, a forma farmacêutica e a quantidade, o nome do medicamento de referência e o respectivo detentor do registro, ficando dispensadas as informações dos artigos 17, 22, 23 e 27 deste Regulamento. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 13 É permitido somente às distribuidoras de medicamentos, farmácias e drogarias receberem catálogo de produtos contendo as seguintes informações: nome comercial dos medicamentos, incluindo àqueles sujeitos à retenção de receita; a substância ativa de acordo com a DCB/DCI; a apresentação, incluindo a concentração, forma farmacêutica e quantidade; o número de registro na Agência Nacional de Vigilância Sanitária; e o respectivo preço, ficando dispensadas as informações dos artigos 22, 23 e 27. Art. 13 É permitido somente às distribuidoras de medicamentos, farmácias e drogarias receberem catálogo de produtos contendo as seguintes informações: nome comercial dos medicamentos, incluindo àqueles sujeitos à retenção de receita; a substância ativa de acordo com a DCB/DCI; a apresentação, incluindo a concentração, forma farmacêutica e quantidade; o número de registro na Agência Nacional de Vigilância Sanitária; e o respectivo preço, ficando dispensadas as informações dos artigos 17, 22, 23 e 27. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) 346
347 Art. 14 A propaganda ou publicidade de medicamentos não pode utilizar designações, símbolos, figuras ou outras representações gráficas, ou quaisquer indicações que possam tornar a informação falsa, incorreta, ou que possibilitem interpretação falsa, equívoco, erro e/ou confusão em relação à verdadeira natureza, composição, procedência, qualidade, forma de uso, finalidade e/ou características do produto. Art. 15 As comparações realizadas de forma direta ou indireta entre quaisquer medicamentos, isentos de prescrição ou não, devem estar baseadas em informações extraídas de estudos comparativos, veiculados em publicações científicas, preferencialmente com níveis de evidência I ou II, e especificar a referência bibliográfica completa. Parágrafo único. As comparações relacionadas à biodisponibilidade e à bioequivalência de princípios ativos poderão ser feitas com base em estudos emitidos por laboratórios oficiais e aprovados pela Anvisa, desde que devidamente referenciados e disponíveis no site da empresa e SAC. Parágrafo único - As comparações relacionadas à biodisponibilidade e à bioequivalência de princípios ativos poderão ser feitas com base em estudos aprovados pela Anvisa emitidos por laboratórios certificados, desde que devidamente referenciados. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 16 Quando se tratar de medicamento genérico, de acordo com a Lei nº 9.787/99 e suas regulamentações, a propaganda ou publicidade deve incluir a frase: "Medicamento Genérico - Lei nº 9.787/99". Art. 17 A propaganda ou publicidade de medicamentos que apresentem efeitos de sedação e/ou sonolência, conforme a bula domedicamento registrada na Anvisa, deve apresentar a advertência: "(nome comercial do medicamento ou, no caso dos medicamentos genéricos, a substância ativa) é um medicamento. Durante seu uso, não dirija veículos ou opere máquinas, pois sua agilidade e atenção podem estar prejudicadas.", ficando dispensada a advertência do artigo 23 deste Regulamento. Parágrafo único. A advertência a que se refere o caput desse artigo deverá obedecer aos critérios do artigo 23. Parágrafo único. A advertência a que se refere o caput desse artigo deverá obedecer aos critérios do artigo 24. (Redação dada pela RETIFICAÇÃO publicada no DOU de 16/07/2009) Art. 18 Os preços dos medicamentos, quando informados ao público em geral, devem ser indicados por meio de listas nas quais devem constar somente o nome comercial do produto; a substância ativa, segundo a DCB/DCI; a apresentação, incluindo a concentração, forma farmacêutica e a quantidade; o número de registro na Agência Nacional de Vigilância Sanitária; o nome do detentor do registro; e o preço dos medicamentos listados. Parágrafo único. No caso dos medicamentos isentos de prescrição médica, ficam permitidas outras formas de comunicação, que não sejam as listas, desde que incluam as demais informações exigidas por este Regulamento. (Vide a INSTRUÇÃO NORMATIVA Nº 05, DE 20 DE MAIO DE 2009) 347
348 Art. 19 Quando as farmácias e drogarias utilizarem frases para informar a redução de preços para grupos de medicamentos, tais como "desconto para anticoncepcionais", "genéricos com 30% de desconto", não podem ser utilizados outros argumentos de cunho publicitário. Art. 20 Na propaganda ou publicidade dirigida aos profissionais habilitados a dispensar ou prescrever medicamentos, as informações referentes ao preço máximo ao consumidor devem mencionar a respectiva fonte, bem como informar apresentação, incluindo concentração, forma farmacêutica e quantidade do medicamento. Art. 21 No caso específico de ser apresentado o nome e/ou imagem de profissional de saúde, como respaldo das propriedades anunciadas do medicamento, é obrigatório constar, de maneira clara, na mensagem publicitária, o nome do profissional interveniente e seu número de inscrição no respectivo Conselho ou outro órgão de registro profissional. TÍTULO II - REQUISITOS PARA A PROPAGANDA OU PUBLICIDADE DE MEDICAMENTOS INDUSTRIALIZADOS ISENTOS DE PRESCRIÇÃO Art. 22 A propaganda ou publicidade de medicamentos isentos de prescrição médica deve cumprir os requisitos gerais, sem prejuízo do que, particularmente, se estabeleça para determinados tipos de medicamentos, sendo exigido constar as seguintes informações: I - nome comercial do medicamento, quando houver; II - nome da substância ativa de acordo com a DCB e, na sua falta, a DCI ou nomenclatura botânica, que deverá ter, no mínimo, 50% do tamanho do nome comercial; III - número de registro na Anvisa, contemplando, no mínimo, nove dígitos, com exceção das peças publicitárias veiculadas em rádio; IV - no caso dos medicamentos de notificação simplificada, a seguinte frase: "MEDICAMENTO DE NOTIFICAÇÃO SIMPLIFICADA RDC Anvisa Nº.../2006. AFE nº:...", com exceção das peças publicitárias veiculadas em rádio; V - as indicações; VI - data de impressão das peças publicitárias; VII - a advertência: "SE PERSISTIREM OS SINTOMAS, O MÉDICO DEVERÁ SER CONSULTADO", que deve observar o artigo 6º. a) Os requisitos dos incisos "II", "V", "VI" e "VII" aplicam-se às formulações oficinais, tendo como embasamento técnicºcientífico a literatura nacional e internacional, oficialmente reconhecida e relacionada no anexo II deste Regulamento. b) A emissora de rádio, a partir da venda do espaço promocional, deve ter à disposição do consumidor e da autoridade sanitária, a informação sobre o número de registro ou, no caso dos medicamentos de notificação simplificada, a Resolução que autoriza a fabricação, importação e/ou comercialização do medicamento. c) Quando direcionada ao público em geral, os termos técnicos da propaganda ou publicidade de medicamentos isentos de prescrição médica deverão ser escritos de maneira a facilitar a compreensão do público. d) No caso de medicamentos com mais que dois e até quatro substâncias ativas, a veiculação dos nomes das substâncias ativas na propaganda ou publicidade pode ser feita com, no mínimo, 30% do tamanho do nome comercial. (Alínea incluída pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) 348
349 e) No caso de medicamentos com mais de quatro fármacos que tenham algum impedimento técnico de cumprir o disposto no item imediatamente anterior, pode ser utilizado na propaganda ou publicidade o nome genérico do fármaco/ substância ativa que justifique a indicação terapêutica do produto seguida da expressão "+ ASSOCIAÇÃO", em tamanho correspondente a 50% do tamanho do nome comercial. (Alínea incluída pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) f) No caso de complexos vitamínicos e ou minerais, e ou de aminoácidos pode ser utilizado na propaganda ou publicidade as expressões Polivitamínico e ou, Poliminerais e ou Poliaminoácidos, como designação genérica, correspondendo a 50% do tamanho do nome comercial do produto. (Alínea incluída pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 23 A propaganda ou publicidade de medicamentos isentos de prescrição médica deve, também, veicular advertência relacionada à substância ativa do medicamento, conforme tabela do anexo III. Parágrafo único. No caso de não ser contemplada alguma substância ativa ou associação na tabela do anexo III, a propaganda ou publicidade deve veicular a seguinte advertência: "(nome comercial do medicamento ou, no caso dos medicamentos genéricos, a substância ativa) É UM MEDICAMENTO. SEU USO PODE TRAZER RISCOS. PROCURE O MÉDICO E O FARMACÊUTICO. LEIA A BULA". Art. 24 A advertência a que se refere o artigo 23 deve ser contextualizada na peça publicitária, de maneira que seja pronunciada pelo personagem principal, quando veiculada na televisão; proferida pelo mesmo locutor, quando veiculada em rádio; e, quando impressa, deve causar o mesmo impacto visual que as demais informações presentes na peça publicitária, apresentandºse com, no mínimo, 35% do tamanho da maior fonte utilizada. I - A locução das advertências de que trata o caput deste artigo deve ser cadenciada, pausada e perfeitamente audível. II - Se a propaganda ou publicidade de televisão não apresentar personagem principal, as advertências devem observar os seguintes requisitos: a) após o término da mensagem publicitária, a advertência será exibida em cartela única, com fundo azul, em letras brancas, de forma a permitir a perfeita legibilidade e visibilidade, permanecendo imóvel no vídeo; b) a locução deve ser diferenciada, cadenciada, pausada e perfeitamente audível; c) a cartela obedecerá ao gabarito RTV de filmagem no tamanho padrão de 36,5cmx27cm (trinta e seis e meio centímetros por vinte e sete centímetros); d) as letras apostas na cartela serão da família tipográfica Humanist 777 Bold ou Frutiger 55 Bold, corpo 38, caixa alta. III - Na internet, a advertência deve ser exibida permanentemente e de forma visível, inserida em retângulo de fundo branco, emoldurada por filete interno, em letras de cor preta, padrão Humanist 777 Bold ou Frutiger 55 Bold, caixa alta, respeitando a proporção de dois décimos do total do espaço da propaganda. Art. 25 Fica proibida a veiculação, na televisão, de propaganda ou publicidade de medicamentos nos intervalos dos programas destinados a crianças ou adolescentes, conforme classificação do Estatuto da Criança e do Adolescente, bem como em revistas de conteúdo dedicado a este público. 349
350 Art. 25 Fica proibida a veiculação, na televisão, de propaganda ou publicidade de medicamentos nos intervalos dos programas destinados a crianças, conforme classificação do Estatuto da Criança e do Adolescente, bem como em revistas de conteúdo dedicado a este público. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 26 Na propaganda ou publicidade de medicamentos isentos de prescrição é vedado: I - usar expressões tais como: "Demonstrado em ensaios clínicos", "Comprovado cientificamente"; II - sugerir que o medicamento é a única alternativa de tratamento, fazendo crer que são supérfluos os hábitos de vida saudáveis e/ou a consulta ao médico; II - sugerir que o medicamento é a única alternativa de tratamento e/ou fazer crer que são supérfluos os hábito de vida saudáveis e/ou a consulta ao médico; (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) III - apresentar nome, imagem e/ou voz de pessoa leiga em medicina ou farmácia, cujas características sejam facilmente reconhecidas pelo público em razão de sua celebridade, afirmando ou sugerindo que utiliza o medicamento ou recomendando o seu uso; IV - usar de linguagem direta ou indireta relacionando o uso de medicamento a excessos etílicos ou gastronômicos; V - usar de linguagem direta ou indireta relacionando o uso de medicamento ao desempenho físico, intelectual, emocional, sexual ou à beleza de uma pessoa, exceto quando forem propriedades aprovadas pela Anvisa; VI - apresentar de forma abusiva, enganosa ou assustadora representações visuais das alterações do corpo humano causadas por doenças ou lesões; VII - incluir mensagens, símbolos e imagens de qualquer natureza dirigidas a crianças ou adolescentes, conforme classificação do Estatuto da Criança e do Adolescente. TÍTULO III - REQUISITOS PARA PROPAGANDA OU PUBLICIDADE DE MEDICAMENTOS INDUSTRIALIZADOS DE VENDA SOB PRESCRIÇÃO Art. 27 A propaganda ou publicidade de medicamentos de venda sob prescrição deve cumprir os requisitos gerais, sem prejuízo do que, particularmente, se estabeleça para determinados tipos de medicamentos, e fica restrita aos meios de comunicação destinados exclusivamente aos profissionais de saúde habilitados a prescrever ou dispensar tais produtos, devendo incluir informações essenciais referentes: I - ao nome comercial do medicamento, quando houver; II - ao nome da substância ativa de acordo com a DCB e, na sua falta, a DCI ou nomenclatura botânica, que deverá ter, no mínimo, 50% do tamanho do nome comercial; III - ao número de registro na Anvisa, contemplando no mínimo os nove dígitos; IV - às indicações; V - às contra-indicações; VI - aos cuidados e advertências (contemplando as reações adversas e interações com medicamentos, alimentos e álcool); VII - à posologia; VIII - à classificação do medicamento em relação à prescrição e dispensação; IX - à data de impressão das peças publicitárias impressas. 350
351 1º As informações exigidas por este artigo devem se apresentar com fonte de, no mínimo, dois milímetros. 2º Na propaganda ou publicidade de vacinas, deverá constar, ainda, a informação sobre o número de doses necessárias para uma completa imunização. Art. 28 Na propaganda ou publicidade de medicamentos de venda sob prescrição, quando forem destacados os benefícios do medicamento no texto da peça publicitária, devem ser destacadas, pelo menos, uma contra-indicação e uma interação medicamentosa mais freqüente, dentre aquelas exigidas no artigo 27, incisos, V e VI, causando impacto visual ao leitor e obedecendo à proporcionalidade de 20% do tamanho da maior fonte utilizada. Art. 29 A propaganda ou publicidade de medicamentos de venda sob prescrição veiculada na internet deve ser acessível, exclusivamente, aos profissionais habilitados a prescrever ou dispensar medicamentos, por meio de sistema de cadastramento eletrônico, devendo ser apresentado um termo de responsabilidade informando sobre a restrição legal do acesso. Parágrafo único. As bulas dos medicamentos de venda sob prescrição médica veiculadas na internet, sem acesso restrito, devem ser atualizadas, reproduzir fielmente as aprovadas pela Anvisa e não podem apresentar designações, símbolos, figuras, desenhos, imagens, slogans e quaisquer argumentos de cunho publicitário em relação aos medicamentos. Art. 30 Quaisquer afirmações, citações, tabelas ou ilustrações relacionadas a informações científicas devem ser extraídas de estudos clínicos, veiculados em publicações científicas, preferencialmente com níveis de evidência I ou II. 1º As afirmações, citações, tabelas ou outras ilustrações a que se refere o caput do artigo devem ser fielmente reproduzidas e especificar a referência bibliográfica. 2º A criação de gráficos, quadros, tabelas e ilustrações de mecanismos de ação para transmitir informações, que não estejam assim representadas nos estudos científicos, deve expressar com rigor a veracidade das informações e especificar a referência bibliográfica completa. 3º Os gráficos, tabelas e ilustrações de mecanismos de ação de que trata este artigo devem ser verdadeiros, exatos, completos, não tendenciosos, bem como não podem ser apresentados de forma que possibilitem erro ou confusão quanto às características do medicamento através do impacto visual. Art. 31 As afirmações relacionadas à biodisponibilidade e à bioequivalência de princípios ativos poderão ser feitas com base em estudos emitidos por emitidos por laboratórios habilitados e aprovados na Anvisa, desde que devidamente referenciados e disponíveis no site da empresa e no SAC. Art. 31 As afirmações relacionadas à biodisponibilidade e à bioequivalência de princípios ativos poderão ser feitas com base em estudos aprovados pela Anvisa emitidos por laboratórios certificados, desde que devidamente referenciados (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 32 A propaganda ou publicidade de medicamentos sob controle especial, sujeitos à venda sob prescrição médica, com notificação de receita ou retenção de receita, além de observar as disposições deste regulamento técnico, somente pode ser efetuada em revistas de conteúdo exclusivamente técnico, referentes a patologias e medicamentos, dirigidas direta e unicamente a profissionais de saúde habilitados a prescrever e/ou dispensar medicamentos. 351
352 1 Ficam excluídas das revistas mencionadas no caput deste artigo, aquelas que possuam matérias de cunho sociocultural e outras que não sejam técnicºcientíficas. 2 É permitida a veiculação de propaganda ou publicidade dos medicamentos citados no caput deste artigo, em cópia fiel de artigo técnicºcientífico referente à substância ativa do medicamento divulgado e publicado em revistas mencionadas no caput, especificando a referência bibliográfica completa, bem como em material de ajuda visual de uso exclusivo do propagandista e monografias do medicamento. TÍTULO IV - REQUISITOS PARA AMOSTRAS GRÁTIS Art. 33 A distribuição de amostras grátis de medicamentos somente pode ser feita pelas empresas aos profissionais prescritores em ambulatórios, hospitais, consultórios médicos e odontológicos. 1º É vedado distribuição de amostras grátis de medicamentos biológicos. 1º É vedado distribuição de amostras grátis de vacinas. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) 2º É vedada a distribuição de amostras grátis de preparações magistrais. 3º É vedada a distribuição de amostras grátis de medicamentos isentos de prescrição. (Revogado pela RESOLUÇÃO-RDC Nº 60, DE 26 DE NOVEMBRO DE 2009) Art. 34 As amostras grátis de medicamentos de venda sob prescrição médica devem conter 50% do conteúdo da apresentação original registrada na Anvisa e comercializada pela empresa, com exceção dos antibióticos, que deverão ter a quantidade suficiente para o tratamento de um paciente, e dos anticoncepcionais e medicamentos de uso contínuo, que deverão ter a quantidade de 100% do conteúdo da apresentação original registrada na Anvisa e comercializada pela empresa. (Vide a INSTRUÇÃO NORMATIVA Nº 05, DE 20 DE MAIO DE 2009) (Revogado pela RESOLUÇÃO-RDC Nº 60, DE 26 DE NOVEMBRO DE 2009) Art. 35 As embalagens das amostras grátis devem conter a expressão ''AMOSTRA GRÁTIS'' não removível. 1º As embalagens secundárias das amostras grátis não podem veicular designações, símbolos, figuras, imagens, desenhos, slogans e quaisquer argumentos de cunho publicitário, exceto quando aprovado pela Anvisa, para constar na embalagem original. 2º Os dizeres de rotulagem e o layout das amostras grátis não contemplados neste artigo, bem como as bulas, etiquetas e prospectos, devem se apresentar idênticos aos aprovados para constar na embalagem original. 3º O número de registro constante na amostra grátis deve conter os treze dígitos correspondentes à embalagem original, registrada e comercializada, da qual se fez a amostra. 4 Deve constar da rotulagem da amostra grátis o número de lote, e a empresa deve manter atualizado e disponível à Agência Nacional de Vigilância Sanitária o quadro de distribuição de amostras por um período mínimo de dois anos. 5 A distribuição de amostras grátis de medicamentos à base de substâncias sujeitas a controle especial dar-se-á também mediante os dispositivos regulamentados na legislação sanitária vigente. (Revogado pela RESOLUÇÃO-RDC Nº 60, DE 26 DE NOVEMBRO DE 2009) 352
353 TÍTULO V - REQUISITOS PARA MATERIAL INFORMATIVO DE MEDICAMENTOS MANIPULADOS Art Para a divulgação de informações sobre medicamentos manipulados é facultado às farmácias o direito de fornecer, exclusivamente aos profissionais habilitados a prescrever medicamentos, material informativo que contenha somente os nomes das substâncias ativas utilizadas na manipulação de fórmulas magistrais, segundo a sua Denominação Comum Brasileira ou, na sua falta, a Denominação Comum Internacional ou a nomenclatura botânica, bem como as respectivas indicações terapêuticas, fielmente extraídas de literatura especializada e publicações científicas, devidamente referenciadas. Parágrafo único. O material informativo a que se refere o caput desse artigo não pode veicular nome comercial, preço, designações, símbolos, figuras, imagens, desenhos, slogans e quaisquer argumentos de cunho publicitário em relação à substância ativa. Art. 37 É vedado fazer propaganda ou publicidade de empresas em blocos de receituários médicos. TÍTULO VI - REQUISITOS PARA A VISITA DE PROPAGANDISTAS Art. 38 Quando as informações técnicas sobre os medicamentos industrializados e manipulados forem levadas aos profissionais prescritores ou dispensadores por intermédio de propagandistas das empresas, elas deverão ser transmitidas com intuito de promover a prescrição e dispensação do medicamento de forma adequada e condizente com a Política Nacional de Medicamentos. 1º Nas suas ações de propaganda ou publicidade, os propagandistas devem limitar-se às informações científicas e características do medicamento registradas na Anvisa. 2º A visita do propagandista não pode interferir na assistência farmacêutica, nem na atenção aos pacientes, bem como não pode ser realizada na presença de pacientes e seus respectivos acompanhantes, ficando a critério das instituições de saúde a regulamentação das visitas dos propagandistas. TÍTULO VII - REQUISITOS PARA PROPAGANDA OU PUBLICIDADE EM EVENTOS CIENTÍFICOS Art. 39 Nos eventos científicos pode ser distribuído aos profissionais de saúde não habilitados a prescrever ou dispensar medicamentos e aos estudantes da área de saúde material científico contendo o nome comercial do medicamento, a substância ativa e o nome da empresa. Art. 40 O material de propaganda ou publicidade de medicamentos deve ser distribuído aos participantes dos eventos que estiverem com a identificação de sua categoria profissional claramente visível nos crachás. Art. 41 A identificação dos espaços na área de exposição e no interior dos auditórios e similares pode apresentar o nome comercial do medicamento, quando for o caso, juntamente com a respectiva substância ativa e/ou o nome da empresa, podendo ser utilizada a marca figurativa ou mista do produto presente na embalagem aprovada pela Anvisa. Parágrafo único - Fica proibido a utilização de designações, símbolos, figuras, imagens, desenhos, slogans e quaisquer argumentos de cunho publicitário em relação aos medicamentos. (Parágrafo incluído pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) 353
354 Art. 42 Qualquer apoio ou patrocínio, total ou parcial, aos profissionais de saúde para participação em eventos científicos, nacionais ou internacionais, não deve estar condicionado à prescrição, dispensação e/ou propaganda ou publicidade de algum tipo de medicamento. 1º O patrocínio por uma ou mais empresas, de quaisquer eventos, simpósios, congressos, reuniões, conferências e assemelhados, públicos ou privados, seja ele parcial ou total, deve ser exposto com clareza no ato da inscrição dos participantes e nos anais, quando estes existirem. 2º Os palestrantes de qualquer sessão científica que estabeleçam relações com laboratórios farmacêuticos ou tenham qualquer outro interesse financeiro ou comercial devem informar potencial conflito de interesses aos organizadores dos congressos, com a devida indicação na programação oficial do evento e no início de sua palestra, bem como, nos anais, quando estes existirem. Art. 43 Os organizadores de eventos científicos que permitam a propaganda ou publicidade de medicamentos devem informar a Anvisa, com antecedência de três meses, a realização de quaisquer eventos científicos regionais, nacionais e internacionais, contemplando local e data de realização, bem como as categorias de profissionais participantes. (Vide a INSTRUÇÃO NORMATIVA Nº 05, DE 20 DE MAIO DE 2009) TÍTULO VIII - REQUISITOS PARA CAMPANHAS SOCIAIS Art. 44 A divulgação de campanha social deve ter como único objetivo informar ações de responsabilidade social da empresa, não podendo haver menção a nomes de medicamentos, nem publicidade destes produtos, da mesma forma que nenhuma propaganda ou publicidade de medicamentos pode se referir às ações de campanhas sociais da empresa. TÍTULO IX - DISPOSIÇÕES GERAIS Art. 45 Fica estabelecido o prazo de 180 (cento e oitenta) dias, a contar da data de publicação deste regulamento, para as empresas e pessoas físicas responsáveis pela propaganda, publicidade, informação e outras práticas cujo objetivo seja a divulgação, promoção ou comercialização de medicamentos se adequarem às novas disposições deste regulamento. Parágrafo único. Excetuam-se do prazo disposto no caput as amostras grátis, que deverão se adequar no prazo de 360 (trezentos e sessenta) dias, a contar da data de publicação deste regulamento. (Revogado pela RESOLUÇÃO-RDC Nº 60, DE 26 DE NOVEMBRO DE 2009) Art. 46 A concessão de redução no preço de medicamento, bem como a sua aquisição de forma gratuita condicionada ao envio de cupons, cartões ou qualquer outro meio ou material, ou ao fornecimento de quaisquer dados que permitam identificar o paciente, o profissional prescritor, a instituição à qual o profissional está vinculado ou o local da prescrição, fica sob regulamentação da Câmara de Regulação de Medicamentos. Art. 47 Os materiais citados nos artigos 12, 13, caput do artigo 18, 39 e 41 não poderão utilizar designações, símbolos, figuras, imagens, desenhos, marcas figurativas ou mistas, slogans e quaisquer argumentos de cunho publicitário em relação aos medicamentos. 354
355 Art. 47 Os materiais citados nos artigos 12, 13, caput do artigo 18 e 39 não poderão utilizar designações, símbolos, figuras, imagens, desenhos, marcas figurativas ou mistas, slogans e quaisquer argumentos de cunho publicitário em relação aos medicamentos. (Redação dada pela RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) Art. 48 Após a publicação da decisão condenatória que aplicar a sanção de mensagem retificadora, o responsável será notificado para apresentar o plano de mídia da propaganda ou publicidade veiculada de forma irregular e uma proposta de mensagem retificadora com o respectivo plano de mídia provisório. 1º A mensagem retificadora deve contemplar: I - declaração de que a empresa ou pessoa física foi condenada em processo administrativo sanitário, instaurado pela Anvisa e/ou autoridade sanitária local, a divulgar mensagem de retificação e esclarecimento para compensar propaganda ou publicidade de produto sujeito à vigilância sanitária veiculada em desconformidade com a legislação sanitária federal; II - listar as irregularidades, identificadas na propaganda e analisadas no processo administrativo sanitário, que culminaram na aplicação da mensagem retificadora, esclarecendo os erros, equívocos e enganos causados e prestando as informações corretas e completas sobre o produto divulgado; III - No caso de medicamentos isentos de prescrição, veicular a seguinte advertência: "Todo medicamento também oferece riscos. Para evitar danos à sua saúde, informe-se." IV - No caso de medicamentos de venda sob prescrição, informar as contra-indicações, cuidados, advertências, reações adversas e interações medicamentosas do medicamento, bem como veicular a seguinte advertência: "Informações equilibradas e avaliadas criteriosamente são essenciais para a prescrição e o uso racional de medicamentos." 2º O plano de mídia provisório poderá ser modificado e/ou adaptado, assim como poderão ser impostos outros requisitos que levarão em consideração o tipo de produto divulgado, o risco sanitário e o público atingido. Art. 49 A veiculação da mensagem retificadora deve observar os seguintes requisitos: I - Na televisão, a mensagem retificadora deve ser veiculada em texto escrito sobre fundo verde, sem imagens, com letras brancas, padrão Humanist 777 ou Frutiger 55, subindo em rol de caracteres, com locução em "off", cadenciada, sem fundo musical e perfeitamente audível. II - Em rádio, a mensagem retificadora deve ser lida sem fundo musical e com locução cadenciada e perfeitamente audível. III - Nos jornais, revistas, mídia exterior e congêneres, a mensagem retificadora deve ser publicada em fundo branco, emoldurado por filete interno e com letras de cor preta, padrão Humanist 777 ou Frutiger 55. IV - Na Internet, a mensagem retificadora deve ser inserida em fundo branco, emoldurado por filete interno, com letras de cor preta, padrão Humanist 777 ou Frutiger 55. V - Caso o espaço publicitário seja suficiente, a mensagem deve ser veiculada em cartela única, com as letras em tamanho legível. Caso não seja suficiente, a mensagem deve ser exibida seqüencialmente e de forma perfeitamente legível. 355
356 VI - O responsável pode ser notificado para apresentar, no prazo de dez dias contados do recebimento da notificação, prorrogável uma única vez por igual período, modificações na mensagem retificadora e no plano de mídia apresentados para adequá-los aos requisitos impostos de acordo com as normas estabelecidas nesta Resolução. Art. 50 Cumpridos todos os requisitos, o responsável será notificado para proceder à divulgação da mensagem retificadora nos meios de comunicação, devendo, em seguida, comprovar a execução completa do plano de mídia da seguinte forma: I - em relação às mensagens retificadoras veiculadas na televisão e no rádio, deve ser juntada aos autos a nota fiscal discriminada, comprovando que a mensagem foi divulgada nos veículos, horários e freqüências previstos no plano de mídia, bem como a gravação da mensagem veiculada; II - em relação às mensagens retificadoras veiculadas em jornais e revistas, deve ser juntado aos autos um exemplar de cada publicação na qual a mensagem foi divulgada; III - em relação às mensagens retificadoras veiculadas na mídia exterior e congêneres, devem ser juntadas aos autos, além da nota fiscal discriminada, comprovando que a mensagem foi divulgada conforme previsto no plano de mídia, fotos com os negativos da mensagem inserida nos respectivos meios; IV - em relação às mensagens retificadoras veiculadas na Internet, deve ser juntado aos autos documento comprovando que a mensagem foi divulgada nos sítios eletrônicos especificados no plano de mídia, bem como a impressão da página contendo a data. 1º Após a divulgação da mensagem retificadora, seguida da comprovação da execução completa do plano de mídia, será expedido um despacho atestando o regular cumprimento da sanção, com a conseqüente extinção do processo administrativo sanitário. 2º No caso de não cumprimento da sanção de mensagem retificadora, o responsável ficará sujeito às conseqüências e penalidades previstas na legislação sanitária. Art. 51 Durante a apuração do ilícito, quando se tratar de propaganda, publicidade ou informação que representem risco sanitário iminente à saúde pública, pode a entidade sanitária, como medida cautelar, determinar a suspensão da veiculação do material publicitário ou informativo, com a duração necessária para a realização de análises ou outras providências requeridas. Art. 52 As empresas devem informar a todo seu pessoal de comercialização e divulgação de medicamentos, incluindo as agênciasde publicidade, sobre este Regulamento Técnico e as responsabilidades no seu cumprimento. Art. 53 A Anvisa, a qualquer tempo, poderá expedir atos regulamentares relativos à matéria, com o propósito de atualizar a regulamentação sobre a propaganda, publicidade, promoção e informação de produtos sujeitos à vigilância sanitária. Art. 54 Ficam expressamente revogadas a RDC 102/2000, RDC 199/2004, RDC 197/2004 e demais Resoluções que dispõem de forma contrária. Art. 54 Ficam expressamente revogadas a RDC 102, de 30 de novembro de 2000, publicada no DOU nº E, Seção 1, pág.28, de 1º de dezembro de 2000, RDC 199, de 17 de agosto de 2004, publicada no DOU nº 159, Seção 1, pág.82, de 18 de agosto de 2004, republicada no DOU nº 164, Seção 1, pág. 47, de 25 de agosto de 2004, o art.90 da Portaria SVS/MS nº 344, de 12 de maio de 1998, publicada no DOU nº 91 - E, Seção 1, pág. 3, de 15 de maio de 1998, republicada DOU de 1º de fevereiro de 1999 e demais normas que dispõem de forma contrária. 356
357 (Redação dada pela Retificação publicada no DOU de 17/08/2010) ANEXO II - LITERATURAS NACIONAIS E INTERNACIONAIS OFICIALMENTE RECONHECIDAS FARMACOPÉIA BRASILEIRA FARMACOPÉIA ALEMÃ FARMACOPÉIA BRITÂNICA FARMACOPÉIA EUROPÉIA FARMACOPÉIA NÓRDICA FARMACOPÉIA JAPONESA FARMACOPÉIA FRANCESA FARMACOPÉIA AMERICANA E SEU FORMULÁRIO NACIONAL FARMACOPÉIA MEXICANA FARMACOPÉIA PORTUGUESA (Farmacopéia Portuguesa incluída pelas Retificações da RESOLUÇÃO-RDC Nº 23, DE 20 DE MAIO DE 2009) USP NATIONAL FORMULARY MARTINDALE, WILLIAN EXTRA PHARMACOPÉIA DICTIONAIRE VIDAL EDITIONS DU VIDAL REMINGTON FARMÁCIA EDITORIAL MÉDICA PANAMERICANA USP DI INFORMACION DE MEDICAMENTOS USP PHARMACISTS' PHARMACOPEIA FORMULÁRIO NACIONAL HOMEOPATHIE - PHARMACOTECHNIE ET MONOGRAPHIES DES MEDICAMENTES COURANTS VOLUME I E II HOMOEPATHIC PHARMACOPEIA OF INDIA PHARMACOPÉE FRANÇAISE E SUPLEMENTOS THE HOMEOPATHIC PHARMACOPOEIA OF THE UNITED STATES E SUPLEMENTOS ANEXO III - (TABELA) PRINCÍPIO ATIVO 1. Ácido acetilsalicílico 2. Ácido ascórbico (vitamina C) 3. Bicarbonato de sódio ALERTAS PARA USO EM PROPAGANDA Não use este medicamento em caso de gravidez, gastrite ou úlcera do estômago e suspeita de dengue ou catapora. Não use este medicamento em caso de doença grave dos rins. Não use este medicamento se você tem restrição ao consumo de sal, insuficiência dos rins, do coração ou do fígado. 4. Bisacodil Não use este medicamento em caso de doenças intestinais graves. 357
358 5. Cânfora Não use este medicamento em crianças menores de dois anos de idade. 6. Carbonato de Cálcio Não use este medicamento em caso de doença dos rins. 7. Carvão vegetal Não use este medicamento em crianças com diarréia aguda e persistente. 8. Cloridrato de ambroxol 9. Cloridrato de fenilefrina Não use este medicamento em crianças menores de dois anos de idade. Não use este medicamento em caso de doenças do coração, pressão alta e glaucoma. 10. Dipirona sódica Não use este medicamento durante a gravidez e em crianças menores de três meses de idade. 11. Dropropizina Não use este medicamento em caso de tosse com secreção e em crianças menores de dois anos de idade. 12. Hidróxido de alumínio 13. Hidróxido de magnésio Não use este medicamento em caso de doença dos rins e dor abdominal aguda. Não use este medicamento em caso de doença dos rins. 14. Ibuprofeno Não use este medicamento em casos de úlcera, gastrite, doença dos rins ou se você já teve reação alérgica a antiinflamatórios. 15. Mebendazol Não use este medicamento em crianças menores de um ano de idade. 16. Naproxeno. Não use este medicamento em casos de úlcera, gastrite, doença dos rins ou se você já teve reação alérgica a antiinflamatórios. 17. Nicotina Não use este medicamento se você é fumante com problemas cardíacos. 18. Paracetamol Não use junto com outros medicamentos que contenham paracetamol, com álcool, ou em caso de doença grave do fígado. 19. Picossulfato de sódio 20. Plantago ovata Forsk Não use este medicamento em caso de doenças intestinais graves. Não use este medicamento em caso de doenças intestinais graves. 21. Sulfato ferroso Não use este medicamento se você tem problemas gastrointestinais. 358
359 RE Nº 1, DE 29 DE JULHO DE 2005 O Diretor-Presidente da Agência Nacional de Vigilância Sanitária, no uso de suas atribuições e tendo em vista o disposto no art. 111, inciso II, alínea "a" do Regimento Interno da ANVISA aprovado pela Portaria nº 593, de 25 de agosto de 2000, republicada em 22 de dezembro de 2000, resolve: Art. 1º Autorizar ad referendum, a publicação do Guia para a Realização de Estudos de Estabilidade, em anexo. Art. 2 Fica revogada a Resolução - RE n 398, de 12 de novembro de 2004, publicada no Diário Oficial da União de 16 de novembro de Art. 3 Esta Resolução entra em vigor na data de sua publicação. ANEXO - GUIA PARA A REALIZAÇÃO DE ESTUDOS DE ESTABILIDADE A estabilidade de produtos farmacêuticos depende de fatores ambientais como temperatura, umidade e luz, e de outros relacionados ao próprio produto como propriedades físicas e químicas de substâncias ativas e excipientes farmacêuticos, forma farmacêutica e sua composição, processo de fabricação, tipo e propriedades dos materiais de embalagem. APLICABILIDADE Guia para realização dos testes de estabilidade de produtos farmacêuticos a fim de prever, determinar ou acompanhar o seu prazo de validade. 1. DEFINIÇÕES ESTUDO DE ESTABILIDADE ACELERADO Estudo projetado para acelerar a degradação química e/ou mudanças físicas de um produto farmacêutico em condições forçadas de armazenamento. Os dados assim obtidos, juntamente com aqueles derivados dos estudos de longa duração, podem ser usados para avaliar efeitos químicos e físicos prolongados em condições não aceleradas e para avaliar o impacto de curtas exposições a condições fora daquelas estabelecidas no rótulo do produto, que podem ocorrer durante o transporte. ESTUDO DE ESTABILIDADE DE ACOMPANHAMENTO Estudo realizado para verificar que o produto farmacêutico mantém suas características físicas, químicas, biológicas, e microbiológicas conforme os resultados obtidos nos estudos de estabilidade de longa duração. ESTUDO DE ESTABILIDADE DE LONGA DURAÇÃO Estudo projetado para verificação das características físicas, químicas, biológicas e microbiológicas de um produto farmacêutico durante e, opcionalmente, depois do prazo de validade esperado. Os resultados são usados para estabelecer ou confirmar o prazo de validade e recomendar as condições de armazenamento. LOTE 359
360 Quantidade de um produto obtido em um único processo ou série de processos, cujas características essenciais são a homogeneidade e qualidade dentro dos limites especificados. LOTE EM ESCALA PILOTO Um lote de produto farmacêutico produzido por um processo totalmente representativo simulando o lote de produção industrial e estabelecido por uma quantidade mínima equivalente a 10% do lote industrial previsto, ou quantidade equivalente à capacidade mínima do equipamento industrial a ser utilizado. PRAZO DE VALIDADE Data limite para a utilização de um produto farmacêutico definida pelo fabricante, com base nos seus respectivos testes de estabilidade, mantidas as condições de armazenamento e transporte estabelecidos. TESTE DE ESTABILIDADE Conjunto de testes projetados para obter informações sobre a estabilidade de produtos farmacêuticos visando definir seu prazo de validade e período de utilização em embalagem e condições de armazenamento especificadas. 2. DISPOSIÇÕES GERAIS 2.1 O prazo de validade de um produto a ser comercializado no Brasil é determinado por um estudo de estabilidade de longa duração de acordo com os parâmetros definidos em tabela abaixo. Por ocasião do registro poderá ser concedido um prazo de validade provisório de 24 meses se aprovado o relatório de estudo de estabilidade de longa duração de 12 meses ou relatório de estudo de estabilidade acelerado de 6 meses acompanhado dos resultados preliminares do estudo de longa duração com, conforme parâmetros definidos em tabela abaixo. Forma Farmacêutica Sólido 15 C - 30 C Sólido 15 C - 30 C Semi-sólido *** 15 C - 30 C Semi-sólido 15 C - 30 C Líquidos *** 15 C - 30 C Líquidos 15 C - 30 C Gases 15 C - 30 C Condição de Embalagem Temperatura e armazenamento* umidade Acelerado ** Semi-permeável 40ºC ± 2ºC / 75% UR± 5% UR Impermeável 40ºC ± 2ºC 30ºC ± 2ºC Semi-permeável 40ºC ± 2ºC / 75% UR± 5% UR Impermeável 40ºC ± 2ºC 30ºC ± 2ºC Semi-permeável 40ºC ± 2ºC / 75% UR ± 5% UR Impermeável 40ºC ± 2ºC 30ºC ± 2ºC Impermeável 40ºC ± 2ºC 30ºC ± 2ºC Todas as formas 2 C - 8 C Impermeável 25ºC ± 2ºC 5ºC ± 3ºC Temperatura e umidade Longa Duração ** 30ºC ± 2ºC / 75% UR ± 5% UR 30ºC ± 2ºC / 75% UR ± 5% UR 30ºC ± 2ºC / 75% UR ± 5% UR 360
361 farmacêuticas Todas as formas farmacêuticas Todas as formas farmacêuticas 2 C - 8 C Semi-permeável 25ºC ± 2ºC / 60 % UR ± 5% UR 5ºC ± 3ºC -20 C Todas - 20ºC ± 5ºC - 20ºC ± 5ºC * Qualquer recomendação de armazenamento em temperatura dentro destas faixas deve constar de bulas e rótulos. A temperatura recomendada não exime de que os testes de estabilidade sejam realizados com as temperaturas definidas nas duas últimas colunas da tabela. ** Os valores de temperatura e umidade são fixos e as variações são inerentes às oscilações esperadas pela câmara climática e por eventuais aberturas para retirada ou colocação de material. *** Líquidos e semi-sólidos de base aquosa devem realizar o estudo com umidade a 25% UR ou 75% UR. Caso se opte por 75% UR, o valor da perda de peso deverá ser multiplicado por 3, O prazo de validade deve ser confirmado mediante a apresentação de um estudo de estabilidade de longa duração de 24 meses de duração, protocolado na forma de complementação de informações ao processo. A presença desta documentação no processo é necessária para a renovação do registro O estudo de estabilidade deve ser executado com o produto farmacêutico em sua embalagem primária Os produtos importados a granel devem descrever nos seus rótulos a data de fabricação, a validade e a condição de armazenamento até a execução da embalagem primária para serem liberados pela autoridade sanitária de portos e aeroportos. O estudo será avaliado durante a inspeção na empresa fabricante Para produtos importados, os estudos de estabilidade podem ser realizados no exterior de acordo com os parâmetros definidos nesta Resolução. Nos caso de produtos importados a granel, o prazo de validade deve levar em consideração o tempo máximo de armazenamento até a execução da embalagem primária Para produtos importados, a granel ou em embalagem primária, os estudos de estabilidade de acompanhamento devem ser realizados em solo brasileiro de acordo com os parâmetros definidos nesta Resolução Estudos adicionais, tais como fotoestabilidade que se façam pertinentes de acordo com as propriedades do produto em questão, poderão ser necessárias para a comprovação da estabilidade de produtos farmacêuticos. Nestes casos sugerimos seguir recomendação técnica disponível no portal da Anvisa. A não apresentação de estudo de fotoestabilidade deve vir acompanhada de justificativa técnica com evidência científica de que o(s) ativos(s) não sofre(m) degradação em presença de luz ou de que a embalagem primária não permite a passagem de luz É facultado utilizar modelos reduzidos de plano de estudo de estabilidade, baseados nos princípios estabelecidos na recomendação técnica disponível no portal da Anvisa Todo relatório de estudo de estabilidade, independente da forma farmacêutica, deve apresentar as seguintes informações ou justificativa técnica de ausência: Descrição do produto com respectiva especificação da embalagem primária Número do lote para cada lote envolvido no estudo Descrição do fabricante dos princípios ativos utilizados Aparência Plano de estudo: material, métodos (desenho) e cronograma. Data de início do estudo Teor do princípio ativo e método analítico correspondente Quantificação de produtos de degradação e método analítico correspondente 361
362 Limites microbianos Para toda a forma farmacêutica sólida a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: Dissolução Dureza Para as formas farmacêuticas líquídas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: ph Sedimentação pós agitação em suspensões Claridade em soluções Separação de fase em emulsões e cremes Perda de peso em produtos de base aquosa Para fins de prazo de validade provisório de 24 meses será aprovado o relatório de estabilidade acelerado ou de longa duração de 12 meses que apresentar variação menor ou igual a 5,0% do valor de análise da liberação do lote, mantidas as demais especificações. Caso as variações de doseamento estejam entre 5,1% e 10,0% no estudo de estabilidade acelerado, o prazo de validade provisório será reduzido à metade, ou seja, será de 12 meses. O doseamento no momento zero não pode ultrapassar as especificações do produto de acordo com farmacopéias reconhecidas pela Anvisa ou, na ausência de informação farmacopeica, com método validado de acordo com o Guia para validação de métodos analíticos e bioanalíticos. Caso a especificação farmacopeica e/ou proveniente de método validado permitir que o momento zero seja acima de 10% do declarado a variação da queda será analisada caso a caso Para fins de prazo de validade definitivo, somente será aprovado o relatório de estabilidade que apresentar a variação do doseamento dos princípios ativos dentro das especificações farmacopeicas e/ou proveniente de método validado do produto de acordo com o Guia para validação de métodos analíticos e bioanalíticos, e mantidas as demais características do produto Em caso de produtos que requeiram reconstituição ou diluição deve-se apresentar informações iniciais e finais que comprovem o período de utilização pelo qual o produto mantém a sua estabilidade depois da reconstituição, nas condições de armazenamento determinadas. Os estudos devem ser conduzidos utilizando o diluente especificado para reconstituição do produto farmacêutico. Se existir a opção de mais de um diluente, o estudo deve ser conduzido com aquele que apresente o produto farmacêutico reconstituído menos estável Em caso de comprimidos efervescentes deve-se apresentar informações iniciais e finais que comprovem o período de utilização pelo qual o produto remanescente mantém a sua estabilidade depois da abertura da embalagem primária nas condições de armazenamento determinadas. Estes estudos devem ser realizados com os parâmetros e testes definidos por esta Resolução Excepcionalmente, para os produtos cujos cuidados de conservação sejam inferiores a 25 C e de uso exclusivo em hospitais e clínicas médicas, serão aceitos estudos de estabilidade nas condições especificadas para Zona II (25 C/60%UR), desde que seja devidamente comprovado que o produto não suporta as condições estabelecidas nesta Resolução. Entretanto o titular do registro do produto deve assegurar a conservação recomendada durante o transporte e a distribuição. 3. SELEÇÃO DE LOTES 362
363 3.1. Para fins de registro e alterações pós-registro, nos estudos de estabilidade acelerado e longa duração: um ou três lotes, de acordo com as normas legais e regulamentares pertinentes Os lotes a serem amostrados devem ser representativos do processo de fabricação, tanto em escala piloto quanto escala industrial Para os produtos cuja concentração do princípio ativo esteja na ordem de dosagem abaixo de 0.99 miligramas por unidade posológica, não serão permitidos lotes pilotos com quantitativos diferentes dos lotes industriais. Não aplicável a soluções Os estudos de acompanhamento deverão ser realizados nas condições climáticas preconizadas neste Guia. A amostragem deve seguir os parâmetros abaixo descritos: a) Um lote anual, para produção acima de 15 lotes/ano. b) Um lote a cada 2 anos, produção abaixo ou igual de 15 lotes/ano. c) Para produtos com diferentes concentrações e formulações proporcionais, poderá ser utilizado como critério de escolha, aquele que apresentar o maior número de lotes produzidos ao ano O estudo de acompanhamento somente poderá ser realizado se o produto não sofrer nenhuma alteração após a conclusão do estudo de estabilidade de longa duração. Caso ocorra qualquer alteração no produto deverá ser realizado novo estudo de estabilidade de longa duração conforme preconizado neste Guia. 4. FREQÜÊNCIA DOS TESTES 4.1. Estudo acelerado: 0, 3 e 6 meses para doseamento, quantificação de produtos de degradação, dissolução (quando aplicável) e ph (quando aplicável). Para as demais provas apresentar estudo aos 6 meses comparativo ao momento zero Estudo de longa duração: 0, 3, 6, 9, 12, 18, 24 meses para doseamento, quantificação de produtos de degradação, dissolução (quando aplicável) e ph (quando aplicável). Para as demais provas, apresentar estudo no prazo de validade requerido comparativo ao momento zero Estudo de acompanhamento: a cada 12 meses deverão ser realizados todos os testes de um relatório de estudo de estabilidade, relatório que deve ser disponibilizado no momento da inspeção. 5. DA ADEQUAÇÃO 5.1. É obrigatória a apresentação de estudos de estabilidade no momento da primeira renovação de registro após a publicação desta Resolução caso este estudo não conste do dossiê do registro, mesmo que conduzidos de acordo com os parâmetros vigentes quando do início dos estudos A Anvisa aceitará até 31 de julho de 2007, no momento do registro, pós-registro ou da renovação de registro, estudos de estabilidade de longa duração que já estejam em andamento com o parâmetro de umidade abaixo de 75%. Entretanto as câmaras climáticas devem ser re-qualificadas para umidade de 75% a partir da data de publicação desta Resolução. Fica a critério da empresa reiniciar ou não estes estudos Caso os estudos de estabilidade de longa duração tenham sido realizados somente com parâmetros de umidade distintos do definido nesta Resolução, as empresas deverão na primeira renovação de registro após 1 de agosto de 2007, apresentar estudos de estabilidade de acompanhamento em um lote, de acordo com esta Resolução. Para produtos que atendam a esta circunstância, o momento da renovação é entre 1 de agosto de 2007 e 31 de julho de 2008 e já tenham validade igual ou superior a 36 meses, é possível aprovar um período de validade de 36 meses com a apresentação de estudos de no mínimo de 24 meses. 363
364 5.4. Caso os estudos de estabilidade de longa duração tenham sido realizados com parâmetros de temperatura e umidade distintos do definido nesta Resolução, as empresas deverão na primeira renovação de registro após 1 de agosto de 2007, apresentar estudos de estabilidade de longa duração de 12 meses, ou estudo acelerado de 6 meses acompanhado do respectivo estudo de longa duração de acordo com esta Resolução. Caso as condições de estabilidade não forem comprovadas na submissão da renovação de registro, a empresa deverá solicitar suspensão de comercialização para manter o registro, caso contrário o registro não será renovado Casos os estudos de longa duração, realizados através das condições desta Resolução, comprovem um prazo de validade menor que o estabelecido no registro do produto, a empresa deverá imediata e provisoriamente implementar e solicitar alteração pósregistro para alteração de prazo de validade com base nos dados obtidos. 364
365 RESOLUÇÃO-RE Nº 899, DE 29 DE MAIO DE 2003 O Adjunto da Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição, que lhe confere a Portaria nº 238, de 31 de março de 2003, considerando o disposto no art.111, inciso II, alínea "a" 3º do Regimento Interno aprovado pela Portaria nº 593, de 25 de agosto de 2000, republicada no DOU de 22 de dezembro de 2000, considerando que a matéria foi submetida à apreciação da Diretoria Colegiada, que a aprovou em reunião realizada em 6 de março de 2003, resolve: Art. 1º Determinar a publicação do "Guia para validação de métodos analíticos e bioanalíticos" anexo Art. 2º Fica revogada a Resolução RE nº 475, de 19 de março de Art. 3º Esta Resolução entra em vigor na data de sua publicação. ANEXO - GUIA PARA VALIDAÇÃO DE MÉTODOS ANALÍTICOS E BIOANALÍTICOS MÉTODOS ANALÍTICOS 1. Considerações gerais 1.1. As informações contidas nesse Anexo apresentam as características a serem consideradas durante a validação de procedimentos analíticos. O objetivo de uma validação é demonstrar que o método é apropriado para a finalidade pretendida, ou seja, a determinação qualitativa, semi-quantitativa e/ou quantitativa de fármacos e outras substâncias em produtos farmacêuticos Essas informações aplicam-se a: técnicas analíticas que façam uso de métodos de cromatografia gasosa (CG) ou cromatografia líquida de alta eficiência (CLAE); métodos não-cromatográficos, desde que estes ofereçam uma seletividade aceitável (por ex. titulometria, espectrofotometria UV-VIS); testes imunológicos ou microbiológicos, desde que observado o grau de variabilidade usualmente associado a estas técnicas A validação deve garantir, por meio de estudos experimentais, que o método atenda às exigências das aplicações analíticas, assegurando a confiabilidade dos resultados. Para tanto, deve apresentar especificidade, linearidade, intervalo, precisão, sensibilidade, limite de quantificação, exatidão, adequados à análise Deve-se utilizar substâncias de referência oficializadas pela Farmacopéia Brasileira ou, na ausência destas, por outros códigos autorizados pela legislação vigente. No caso da inexistência dessas substâncias, será admitido o uso de padrões de trabalho, desde que a identidade e o teor sejam devidamente comprovados. 365
366 1.5. Para efeito desse guia, considera-se corrida analítica as medições sucessivas de um mesmo analito, efetuadas nas mesmas condições: método, analista, instrumentação, local, condições de utilização e em intervalo de tempo curto entre as medições No caso de metodologia analítica descrita em farmacopéias ou formulários oficiais, devidamente reconhecidos pela ANVISA, a metodologia será considerada validada No caso de metodologia analítica não descrita em farmacopéias ou formulários oficiais, devidamente reconhecidos pela ANVISA, a metodologia será considerada validada, desde que sejam avaliados os parâmetros relacionados a seguir, conforme especificado nas Tabelas 1 e Especificidade e Seletividade Linearidade Intervalo Precisão Limite de detecção (sensibilidade) Limite de quantificação Exatidão Robustez 1.8. No caso da transferência de metodologias da matriz para suas subsidiárias no Brasil e/ou das empresas nacionais para os centro de estudos de equivalência farmacêutica, a metodologia será considerada validada, desde que sejam avaliados os parâmetros de precisão, especificidade e linearidade. Cópia de toda a documentação original da validação da metodologia deverá ser anexada, como prova de que a metodologia foi originalmente validada e deverá conter, no mínimo, todos os parâmetros relacionados no item Para a garantia da qualidade analítica dos resultados, todos os equipamentos utilizados na validação devem estar devidamente calibrados e os analistas devem ser qualificados e adequadamente treinados Os testes são classificados em 4 categorias, conforme a Tabela 1. TABELA 1. CLASSIFICAÇÃO DOS TESTES, SEGUNDO SUA FINALIDADE Categoria Finalidade do teste I II III IV Testes quantitativos para a determinação do princípio ativo em produtos farmacêuticos ou matérias-primas Testes quantitativos ou ensaio limite para a determinação de impurezas e produtos de degradação em produtos farmacêuticos e matérias-primas Testes de performance (por exemplo: dissolução, liberação do ativo) Testes de identificação Para cada categoria será exigido um conjunto de testes, relacionados na Tabela
367 TABELA 2. ENSAIOS NECESSÁRIOS PARA A VALIDAÇÃO DO MÉTODO ANALÍTICO, SEGUNDO SUA FINALIDADE Parâmetro Categoria I Categoria II Quantitativo Ensaio limite Categoria III Categoria IV Especificidade Sim Sim Sim * Sim Linearidade Sim Sim Não * Não Intervalo Sim Sim * * Não Precisão Repetibilidad e Sim Sim Não Sim Não Intermediária ** ** Não ** Não Limite de detecção Não Não Sim * Não Limite de quantificação Não Sim Não * Não Exatidão Sim Sim * * Não Robustez Sim Sim Sim Não Não * pode ser necessário, dependendo da natureza do teste específico. ** se houver comprovação da reprodutibilidade não é necessária a comprovação da Precisão Intermediária metodologia analítica deverá ser revalidada nas seguintes circunstâncias: mudanças na síntese da substância ativa; mudanças na composição do produto acabado; mudanças no procedimento analítico. Determinadas outras mudanças podem requerer validação também, dependendo da natureza das mudanças. 2. Metodologia 2.1. Especificidade e Seletividade É a capacidade que o método possui de medir exatamente um composto em presença de outros componentes tais como impurezas, produtos de degradação e componentes da matriz Para análise qualitativa (teste de identificação) é necessário demonstrar a capacidade de seleção do método entre compostos com estruturas relacionadas que podem estar presentes. Isto deve ser confirmado pela obtenção de resultados positivos (preferivelmente em relação ao material de referência conhecido) em amostras contendo o fármaco, comparativamente com resultados negativos obtidos com amostras que não contém o fármaco, mas compostos estruturalmente semelhantes Para análise quantitativa (teor) e análise de impurezas, a especificidade pode ser determinada pela comparação dos resultados obtidos de amostras (fármaco ou medicamento) contaminadas com quantidades apropriadas de 367
368 impurezas ou excipientes e amostras não contaminadas, para demonstrar que o resultado do teste não é afetado por esses materiais. Quando a impureza ou o padrão do produto de degradação não estiverem disponíveis, pode-se comparar os resultados do teste das amostras contendo impurezas ou produtos de degradação com os resultados de um segundo procedimento bem caracterizado (por exemplo metodologia farmacopéica ou outro procedimento validado). Estas comparações devem incluir amostras armazenadas sob condições de estresse (por ex. luz, calor umidade, hidrólise ácida/ básica, oxidação) Em métodos cromatográficos, deve-se tomar as precauções necessárias para garantir a pureza dos picos cromatográficos. A utilização de testes de pureza de pico (por exemplo, com auxilio de detector de arranjo de fotodiodos ou espectrometria de massas) são interessantes para demonstrar que o pico cromatográfico é atribuído a um só componente Linearidade É a capacidade de uma metodologia analítica de demonstrar que os resultados obtidos são diretamente proporcionais à concentração do analito na amostra, dentro de um intervalo especificado Recomenda-se que a linearidade seja determinada pela análise de, no mínimo, 5 concentrações diferentes. Estas concentrações devem seguir os intervalos da Tabela Se houver relação linear aparente após exame visual do gráfico, os resultados dos testes deverão ser tratados por métodos estatísticos apropriados para determinação do coeficiente de correlação, intersecção com o eixo Y, coeficiente angular, soma residual dos quadrados mínimos da regressão linear e desvio padrão relativo. Se não houver relação linear, realizar transformação matemática O critério mínimo aceitável do coeficiente de correlação (r) deve ser = 0, Deve-se apresentar as curvas obtidas (experimental e a resultante do tratamento matemático) Intervalo O intervalo especificado é a faixa entre os limites de quantificação superior e inferior de um método analítico. Normalmente é derivado do estudo de linearidade e depende da aplicação pretendida do método (Tabela 3). É estabelecido pela confirmação de que o método apresenta exatidão, precisão e linearidade adequados quando aplicados a amostras contendo quantidades de substâncias dentro do intervalo especificado. TABELA 3. LIMITES PORCENTUAIS DO TEOR DO ANALITO QUE DEVEM ESTAR CONTIDOS NO INTERVALO DE LINEARIDADE PARA ALGUNS MÉTODOS ANALITICOS. Ensaio Alcance Determinação De 80% a 120% da concentração teórica do teste quantitativa do analito em matérias-primas ou em formas farmacêuticas Determinação impurezas de Do nível de impureza esperado até 120% do limite máximo especificado. Quando apresentarem importância toxicológica ou efeitos farmacológicos inesperados, os limites de quantificação e detecção devem ser adequados às 368
369 Uniformidade conteúdo Ensaio de dissolução 2.4. Precisão quantidades de impurezas a serem controladas de De 70% a 130% da concentração teórica do teste De ±?20% sobre o valor especificado para o intervalo. Caso a especificação para a dissolução envolva mais que um tempo, o alcance do método deve incluir -20% sobre o menor valor e +20% sobre o maior valor. A precisão é a avaliação da proximidade dos resultados obtidos em uma série de medidas de uma amostragem múltipla de uma mesma amostra. Esta é considerada em três níveis Repetibilidade (precisão intra-corrida): concordância entre os resultados dentro de um curto período de tempo com o mesmo analista e mesma instrumentação. A repetibilidade do método é verificada por, no mínimo, 9 (nove) determinações, contemplando o intervalo linear do método, ou seja, 3 (três) concentrações, baixa, média e alta, com 3 (três) réplicas cada ou mínimo de 6 determinações a 100% da concentração do teste; Precisão intermediária (precisão inter-corridas): concordância entre os resultados do mesmo laboratório, mas obtidos em dias diferentes, com analistas diferentes e/ou equipamentos diferentes. Para a determinação da precisão intermediária recomenda-se um mínimo de 2 dias diferentes com analistas diferentes Reprodutibilidade (precisão inter-laboratorial): concordância entre os resultados obtidos em laboratórios diferentes como em estudos colaborativos, geralmente aplicados à padronização de metodologia analítica, por exemplo, para inclusão de metodologia em farmacopéias. Estes dados não precisam ser apresentados para a concessão de registro. A precisão de um método analítico pode ser expressa como o desvio padrão ou desvio padrão relativo (coeficiente de variação) de uma série de medidas. A precisão pode ser expressa como desvio padrão relativo (DPR) ou coeficiente de variação (CV%), segundo a fórmula, FÓRMULA em que, DP é o desvio padrão e CMD, a concentração média determinada. O valor máximo aceitável deve ser definido de acordo com a metodologia empregada, a concentração do analito na amostra, o tipo de matriz e a finalidade do método, não se admitindo valores superiores a 5%. 369

370 2.5. Limite de Detecção Limite de detecção é a menor quantidade do analito presente em uma amostra que pode ser detectado, porém não necessariamente quantificado, sob as condições experimentais estabelecidas O limite de detecção é estabelecido por meio da análise de soluções de concentrações conhecidas e decrescentes do analito, até o menor nível detectável; No caso de métodos não instrumentais (CCD, titulação, comparação de cor), esta determinação pode ser feita visualmente, onde o limite de detecção é o menor valor de concentração capaz de produzir o efeito esperado (mudança de cor, turvação, etc) No caso de métodos instrumentais (CLAE, CG, absorção atômica), a estimativa do limite de detecção pode ser feita com base na relação de 3 vezes o ruído da linha de base. Pode ser determinado pela equação, FÓRMULA em que: DPa é o desvio padrão do intercepto com o eixo do Y de, no mínimo, 3 curvas de calibração construídas contendo concentrações do fármaco próximas ao suposto limite de quantificação. Este desvio padrão pode ainda ser obtido a partir da curva de calibração proveniente da análise de um número apropriado de amostras do branco; IC é a inclinação da curva de calibração Limite de Quantificação É a menor quantidade do analito em uma amostra que pode ser determinada com precisão e exatidão aceitáveis sob as condições experimentais estabelecidas. O limite de quantificação é um parâmetro determinado, principalmente, para ensaios quantitativos de impurezas, produtos de degradação em fármacos e produtos de degradação em formas farmacêuticas e é expresso como concentração do analito (por exemplo, porcentagem p/p ou p/v, partes por milhão) na amostra O limite de quantificação é estabelecido por meio da análise de soluções contendo concentrações decrescentes do fármaco até o menor nível determinável com precisão e exatidão aceitáveis. Pode ser expresso pela equação, FÓRMULA 370
371 em que: DPa é o desvio padrão do intercepto com o eixo do Y de, no mínimo, 3 curvas de calibração construídas contendo concentrações do fármaco próximas ao suposto limite de quantificação. Este desvio padrão pode ainda ser obtido a partir da curva de calibração proveniente da análise de um apropriado número de amostras do branco; IC é a inclinação da curva de calibração Também pode ser determinado por meio do ruído. Neste caso, determina-se o ruído da linha de base e considera-se como limite de quantificação aquela concentração que produza relação sinal-ruído superior a 10: Exatidão A exatidão de um método analítico é a proximidade dos resultados obtidos pelo método em estudo em relação ao valor verdadeiro. Várias metodologias para a determinação da exatidão estão disponíveis: Fármaco aplicando-se a metodologia analítica proposta na análise de uma substância de pureza conhecida (padrão de referência); comparação dos resultados obtidos com aqueles resultantes de uma segunda metodologia bem caracterizada, cuja exatidão tenha sido estabelecida; Forma Farmacêutica na análise de uma amostra, na qual quantidade conhecida de fármaco foi adicionada a uma mistura dos componentes do medicamento (placebo contaminado); nos casos em que amostras de todos os componentes do medicamento estão indisponíveis, aceita-se a análise pelo método de adição de padrão, no qual adiciona-se quantidades conhecidas do analito (padrão de referência) ao medicamento Impurezas análise pelo método de adição de padrão, no qual adiciona-se quantidades conhecidas de impurezas e/ou produtos de degradação ao medicamento ou ao fármaco; no caso da indisponibilidade de amostras de certas impurezas e/ou produtos de degradação, aceita-se a comparação dos resultados obtidos com um segundo método bem caracterizado (metodologia farmacopéica ou outro procedimento analítico validado). 371
372 A exatidão é calculada como porcentagem de recuperação da quantidade conhecida do analito adicionado à amostra, ou como a diferença porcentual entre as médias e o valor verdadeiro aceito, acrescida dos intervalos de confiança. A exatidão do método deve ser determinada após o estabelecimento da linearidade, do intervalo linear e da especificidade do mesmo, sendo verificada a partir de, no mínimo, 9 (nove) determinações contemplando o intervalo linear do procedimento, ou seja, 3 (três) concentrações, baixa, média e alta, com 3 (três) réplicas cada. A exatidão é expressa pela relação entre a concentração média determinada experimentalmente e a concentração teórica correspondente: FÓRMULA 2.8. Robustez A robustez de um método analítico é a medida de sua capacidade em resistir a pequenas e deliberadas variações dos parâmetros analíticos. Indica sua confiança durante o uso normal. Durante o desenvolvimento da metodologia, deve-se considerar a avaliação da robustez. Constatando-se a susceptibilidade do método à variações nas condições analíticas, estas deverão ser controladas e precauções devem ser incluídas no procedimento. A Tabela 4 relaciona os principais parâmetros que podem resultar em variação na resposta do método. TABELA 4. FATORES QUE DEVEM SER CONSIDERADOS NA DETERMINAÇÃO DA ROBUSTEZ DO MÉTODO ANALÍTICO. Preparo Amostras Espectrofotometria Cromatografia Líquida Cromatografia Gasosa das - Estabilidade das soluções analíticas - Tempo de extração - Variação do ph da solução - Temperatura - Diferentes fabricantes de solventes - Variação do ph da fase móvel - Variação na composição da fase móvel - Diferentes lotes ou fabricantes de colunas - Temperatura - Fluxo da fase móvel - Diferentes lotes ou fabricantes de colunas - Temperatura - Velocidade do gás de arraste MÉTODOS BIOANALÍTICOS 1. Definições Amostra - termo geral que abrange: controles, brancos, amostras processadas e desconhecidas. 372
373 Amostra branco - amostra de uma matriz biológica na qual nenhum analito foi adicionado, utilizada para avaliar a especificidade do método bioanalítico. Amostra de Controle de Qualidade (CQ) - amostra de matriz biológica adicionada do analito, usada para monitorar o desempenho de um método bioanalítico e para avaliar a integridade e validade dos resultados das amostras desconhecidas analisadas numa corrida individual. Amostra processada - extrato final (anterior à análise instrumental) de uma amostra que foi submetida a várias manipulações (ex.: diluição, extração, concentração). Amostra desconhecida - amostra biológica que é objeto de análise. Analito - composto químico específico a ser mensurado, podendo ser o fármaco não-transformado, biomolécula ou seu derivado, metabólito ou produto de degradação em uma matriz biológica. Corrida analítica (ou lote) - conjunto completo de amostras em estudo, com um número apropriado de padrões e CQs para sua validação e que tem sua análise completa nas mesmas condições. Especificidade - habilidade do método bioanalítico de medir e diferenciar o analito de componentes que possam estar presentes na amostra, tais como metabólitos, impurezas, compostos de degradação ou componentes da matriz. Estabilidade - parâmetro que visa determinar se um analito mantém-se quimicamente inalterado numa dada matriz sob condições específicas, em determinados intervalos de tempo. Exatidão - representa o grau de concordância entre os resultados individuais encontrados e um valor aceito como referência. Faixa de quantificação - corresponde a uma faixa de concentração, incluindo o LSQ e o LIQ, que pode ser confiável e reprodutivelmente quantificada com exatidão e precisão, por meio da relação concentração-resposta. Limite de Detecção (LD) - menor concentração de um analito que o procedimento bioanalítico consegue diferenciar confiavelmente do ruído de fundo. Limite Inferior de Quantificação (LIQ) - menor quantidade de um analito numa amostra que pode ser determinada quantitativamente com precisão e exatidão aceitáveis. Limite Superior de Quantificação (LSQ) - maior quantidade de um analito numa amostra que pode ser determinada quantitativamente com precisão e exatidão. Linearidade - corresponde à capacidade do método de fornecer resultados diretamente proporcionais à concentração da substância em exame (analito). Matriz biológica - material distinto de origem biológica, que pode ser amostrado e processado de modo reprodutível. Método - descrição compreensível de todos os procedimentos usados em análises de amostras. Padrão de calibração - matriz biológica a qual foi adicionada uma quantidade conhecida de analito. Os padrões de calibração são usados para construir a curva de calibração, com a qual são determinadas as concentrações do analito nos CQs e nas amostras desconhecidas em estudo. Padrão Interno (PI) - composto, geralmente com características estruturais similares ao analito, adicionado aos padrões de calibração e amostras em concentrações conhecidas e constantes, para facilitar a determinação do analito. Precisão - representa o grau de repetibilidade entre os resultados de análises individuais, quando o procedimento é aplicado diversas vezes numa mesma amostra homogênea, em idênticas condições de ensaio. Recuperação - eficiência de extração de um método analítico, expressa como a porcentagem da quantidade conhecida de um analito, obtida da comparação dos resultados analíticos de amostras branco acrescidas de padrão e submetidas ao processo de extração, com os resultados analíticos de soluções padrão não extraídas. Reprodutibilidade - precisão entre dois laboratórios. Também representa a precisão do método sob as mesmas condições operacionais, num curto período de tempo. Validação parcial - modificação no método bioanalítico validado que não requer a necessidade de uma revalidação total. 373
374 Validação total - estabelecimento de todos os parâmetros de validação de um método bioanalítico, aplicáveis à análise das amostras. 2. Considerações gerais 2.1. As informações contidas neste guia aplicam-se a métodos bioanalíticos, tais como cromatografia gasosa (CG), cromatografia líquida de alta eficiência (CLAE) e estas combinadas com espectrometria de massa (MS) tais como LC-MS, LC-MS-MS, CGMS, CG-MS-MS, utilizados na determinação quantitativa de fármacos e/ou metabólitos em matrizes biológicas, tais como sangue, soro, plasma ou urina. Também se aplica a outras técnicas analíticas, tais como métodos microbiológicos e imunológicos, ou para outras matrizes biológicas, embora, nestes casos, pode-se observar um alto grau de variabilidade A validação deve garantir, por meio de estudos experimentais, que o método atenda às exigências das aplicações analíticas, assegurando a confiabilidade dos resultados. Para tanto, deve apresentar precisão, exatidão, linearidade, limite de detecção e limite de quantificação, especificidade, reprodutibilidade, estabilidade e recuperação adequadas à análise. Desse modo, é importante ressaltar que todos os equipamentos e materiais devem apresentar-se devidamente calibrados e os analistas devem ser qualificados e adequadamente treinados Deve-se utilizar substâncias químicas de referência e /ou padrões biológicos oficializados pela Farmacopéia Brasileira ou por outros códigos autorizados pela legislação vigente. Serão admitidos estudos utilizando padrões secundários desde que seja comprovada sua certificação, na ausência de substâncias químicas de referência e/ou padrões biológicos farmacopéicos Para os estudos de biodisponibilidade relativa/bioequivalência deve-se utilizar padrão interno, sempre que métodos cromatográficos forem utilizados. Deve-se justificar a impossibilidade de sua utilização Deve ser realizada validação total antes da implementação de um método bioanalítico para a quantificação de um fármaco e/ou metabólitos Devem ser realizadas validações parciais quando ocorrerem modificações no método bioanalítico já validado. Os ensaios de validação parcial podem ser desde uma pequena determinação, como a determinação da exatidão e precisão intra-ensaio, até próximo de uma validação total. As mudanças típicas que podem requerer uma validação parcial incluem, entre outras: transferências de métodos entre laboratórios e analistas; mudanças na metodologia analítica, por exemplo, substituição do sistema de detecção; mudança de anticoagulante na coleta das amostras; mudança de matriz, por exemplo, de plasma para urina; mudança no procedimento de preparação da amostra; mudanças relevantes na faixa de concentração; mudanças de instrumentos e/ou "softwares"; demonstração de seletividade do analito na presença de medicações concomitantes; demonstração de seletividade do analito na presença de metabólitos específicos A avaliação da robustez deve ser considerada durante a fase de desenvolvimento do método. Constatando-se suscetibilidade a variações nas condições analíticas, estas deverão ser adequadamente controladas ou precauções deverão ser incluídas no procedimento. Exemplos de variações: estabilidade das soluções analíticas tempo de extração. Variações típicas em cromatografia líquida: influência da variação de ph da fase móvel influência da variação da composição da fase móvel. 374
375 diferentes colunas (diferentes lotes e/ou fabricantes) temperatura velocidade de fluxo. Variações típicas em cromatografia gasosa: diferentes colunas (diferentes lotes e/ou fabricantes); temperatura; velocidade de fluxo. 3. Validação pré - estudo 3.1. Especificidade Deve-se analisar amostras da matriz biológica (sangue, plasma, soro, urina, ou outra) obtidas de seis indivíduos, sendo quatro amostras normais, uma lipêmica e uma hemolisada, sob condições controladas referentes ao tempo, alimentação e outros fatores importantes para o estudo. Cada amostra branco deve ser testada utilizando o procedimento e as condições cromatográficas propostas. Os resultados devem ser comparados com aqueles obtidos com solução aquosa do analito, em concentração próxima ao LIQ Qualquer amostra branco que apresentar interferência significativa no tempo de retenção do fármaco, metabólito ou padrão interno, deve ser rejeitada. Caso uma ou mais das amostras analisadas apresentarem tal interferência, novas amostras de outros seis indivíduos devem ser testadas. Caso uma ou mais das amostras deste grupo apresentarem interferência significativa no tempo de retenção do fármaco, o método deve ser alterado visando eliminá-la Os interferentes podem ser componentes da matriz biológica, metabólitos, produtos de decomposição e medicamentos utilizados concomitantemente ao estudo. A interferência da nicotina, cafeína, produtos de venda isenta de prescrição e metabólitos deve ser considerada sempre que necessário Caso o método seja destinado à quantificação de mais de um fármaco, cada um deve ser injetado separadamente para determinar os tempos de retenção individuais e assegurar que impurezas de um fármaco não interfiram na análise do outro A resposta de picos interferentes no tempo de retenção do fármaco deve ser inferior a 20% da resposta do LIQ. As respostas de picos interferentes no tempo de retenção do fármaco e do padrão interno devem ser inferiores, respectivamente, a 20% e 5% da resposta na concentração utilizada Curva de calibração/linearidade A curva de calibração representa a relação entre a resposta do instrumento e a concentração conhecida do analito. Deve-se gerar uma curva de calibração para cada fármaco e corrida analítica, a qual será usada para calcular a concentração do fármaco nas amostras, utilizando-se a mesma matriz biológica proposta para o estudo. A curva de calibração deve incluir a análise da amostra branco (matriz biológica isenta de padrão do fármaco e do padrão interno), da amostra zero (matriz biológica mais o padrão interno) e de, no mínimo, 6 (seis) amostras contendo padrão do fármaco e padrão interno, contemplando o limite de variação esperado, do LIQ até 120% da concentração mais alta que se pretende analisar Para a determinação da curva de calibração, deve-se analisar amostras extraídas da matriz apropriada, no mínimo 6 (seis) concentrações diferentes. Procedimentos alternativos devem ser justificados, como na obtenção de uma correlação não-linear, em que um maior número de concentrações de padrões serão necessários Os resultados devem ser analisados por métodos estatísticos apropriados como, por exemplo, o cálculo de regressão linear pelo método dos mínimos quadrados. Deve-se apresentar as curvas obtidas (experimental e a resultante do tratamento matemático), o coeficiente de correlação linear, o coeficiente angular e o intercepto da reta Critérios de aceitação da curva de calibração: desvio menor ou igual a 20% (vinte por cento) em relação a concentração nominal para o LIQ; 375

376 desvio menor ou igual a 15 % (quinze por cento) em relação à concentração nominal para as outras concentrações da curva de calibração; no mínimo quatro de seis concentrações da curva de calibração devem cumprir com os critérios anteriores, incluindo o LIQ e a maior concentração da curva de calibração; o coeficiente de correlação linear deve ser igual ou superior a 0, Precisão A repetibilidade do método é verificada utilizando-se, no mínimo, 3 (três) concentrações (baixa, média e alta), contemplando a faixa de variação do procedimento, realizando-se, no mínimo, 5 (cinco) determinações por concentração A precisão deve ser determinada em uma mesma corrida (precisão intra-corrida) e em corridas diferentes (precisão intercorridas) Pode ser expressa como desvio padrão relativo (DPR) ou coeficiente de variação (CV%), não se admitindo valores superiores a 15%, exceto para o LIQ, para o qual se admite valores menores ou iguais a 20%, segundo a fórmula: FÓRMULA onde, D P é o desvio padrão e C M D, a concentração média determinada Exatidão A exatidão do método deve ser determinada utilizando-se, no mínimo, 3 (três) concentrações (baixa, média e alta), contemplando a faixa de variação do procedimento, realizando-se, no mínimo, 5 (cinco) determinações por concentração A exatidão deve ser determinada em uma mesma corrida analítica (exatidão intra-corrida) e em corridas diferentes (exatidão inter-corridas) O desvio não deve exceder 15%, exceto para o limite de quantificação, para o qual se admite desvios menores ou iguais a 20% A exatidão é expressa pela relação entre a concentração média determinada experimentalmente e a concentração teórica correspondente: FÓRMULA 3.5. Limite inferior de quantificação (LIQ) 376
Agência Nacional de Vigilância Sanitária REGISTRO DE MEDICAMENTOS
 REGISTRO DE MEDICAMENTOS GERÊNCIA GERAL DE MEDICAMENTOS - SETORES A Gerência Geral de Medicamentos está em fase de reestruturação, portanto as divisões setoriais são provisórias; Gerência de Pesquisas,
REGISTRO DE MEDICAMENTOS GERÊNCIA GERAL DE MEDICAMENTOS - SETORES A Gerência Geral de Medicamentos está em fase de reestruturação, portanto as divisões setoriais são provisórias; Gerência de Pesquisas,
RESOLUÇÃO - RDC NO -24, DE 14 DE JUNHO DE 2011
 RESOLUÇÃO - RDC NO -24, DE 14 DE JUNHO DE 2011 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere
RESOLUÇÃO - RDC NO -24, DE 14 DE JUNHO DE 2011 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere
RESOLUÇÃO RDC Nº 59, DE 10 DE OUTUBRO DE 2014. Dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos.
 RESOLUÇÃO RDC Nº 59, DE 10 DE OUTUBRO DE 2014 Dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos. A Diretoria Colegiada da Agência Nacional de Vigilância
RESOLUÇÃO RDC Nº 59, DE 10 DE OUTUBRO DE 2014 Dispõe sobre os nomes dos medicamentos, seus complementos e a formação de famílias de medicamentos. A Diretoria Colegiada da Agência Nacional de Vigilância
Diário Oficial Imprensa Nacional
 Diário Oficial Imprensa Nacional REPÚBLICA FEDERATIVA DO BRASIL BRASÍLIA - DF Nº 116 17/06/11 Seção 1 - p.79 MINISTÉRIO DA SAÚDE AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO - RDC Nº 24, DE 14 DE
Diário Oficial Imprensa Nacional REPÚBLICA FEDERATIVA DO BRASIL BRASÍLIA - DF Nº 116 17/06/11 Seção 1 - p.79 MINISTÉRIO DA SAÚDE AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO - RDC Nº 24, DE 14 DE
Adota a seguinte Consulta Pública e eu, Diretora-Presidente Substituta, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 14, de 16 de março de 2011. D.O.U de 21/03/2011 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 14, de 16 de março de 2011. D.O.U de 21/03/2011 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
RESOLUÇÃO - RDC Nº 132, DE 29 DE MAIO DE 2003(*) Republicada no D.O.U de 02/10/2003
 Republicada no D.O.U de 02/10/2003") RESOLUÇÃO - RDC Nº 132, DE 29 DE MAIO DE 2003(*) Republicada no D.O.U de 02/10/2003 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária,
RESOLUÇÃO - RDC Nº 132, DE 29 DE MAIO DE 2003(*) Republicada no D.O.U de 02/10/2003 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária,
MEDICAMENTOS SIMILARES
 MEDICAMENTOS SIMILARES Fica assegurado o direito de registro de medicamentos similares a outros já registrados, desde que satisfaçam as exigências estabelecidas nesta Lei. (Art. 21 da Lei 6360/76) MEDICAMENTOS
MEDICAMENTOS SIMILARES Fica assegurado o direito de registro de medicamentos similares a outros já registrados, desde que satisfaçam as exigências estabelecidas nesta Lei. (Art. 21 da Lei 6360/76) MEDICAMENTOS
considerando a necessidade de implementar ações que venham contribuir para a melhoria da qualidade da assistência à saúde;
 Resolução - RDC nº 132, de 29 de maio de 2003 D.O.U de 02/06/2003 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária no uso da atribuição
Resolução - RDC nº 132, de 29 de maio de 2003 D.O.U de 02/06/2003 Dispõe sobre o registro de medicamentos específicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária no uso da atribuição
adota a seguinte Consulta Pública e eu, Diretor Presidente, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 19, de 6 de maio de 2009. D.O.U de 11/05/09 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 19, de 6 de maio de 2009. D.O.U de 11/05/09 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das
RDC 60. Perguntas e Respostas. RDC nº 60, RDC 60 - PERGUNTAS E RESPOSTAS
 Regulamentação SOBRE AMOSTRAS GRÁTIS DE MEDICAMENTOS RDC 60 Perguntas e Respostas RDC nº 60, de 26 de NOVEmbro de 2009 1 Regulamentação SOBRE AMOSTRAS GRÁTIS RDC 60 Perguntas e Respostas RDC nº 60, de
Regulamentação SOBRE AMOSTRAS GRÁTIS DE MEDICAMENTOS RDC 60 Perguntas e Respostas RDC nº 60, de 26 de NOVEmbro de 2009 1 Regulamentação SOBRE AMOSTRAS GRÁTIS RDC 60 Perguntas e Respostas RDC nº 60, de
Novas Regras para Rotulagem Medicamentos
 XV ENCONTRO TÉCNICO E XI ENCONTRO EMPRESARIAL - ABRASP Novas Regras para Rotulagem Medicamentos RESOLUÇÃO-RDC Nº 71/2009 Carolina K. Rodrigues 21/09/2010 RESOLUÇÃO-RDC Nº 71/2009 DOU de 23/12/2009 Estabelece
XV ENCONTRO TÉCNICO E XI ENCONTRO EMPRESARIAL - ABRASP Novas Regras para Rotulagem Medicamentos RESOLUÇÃO-RDC Nº 71/2009 Carolina K. Rodrigues 21/09/2010 RESOLUÇÃO-RDC Nº 71/2009 DOU de 23/12/2009 Estabelece
Informe Técnico n. 67, de 1º de setembro de 2015.
 Informe Técnico n. 67, de 1º de setembro de 2015. Assunto: Orientações sobre os procedimentos para solicitação de alterações na lista de alimentos alergênicos. I. Introdução. A Resolução de Diretoria Colegiada
Informe Técnico n. 67, de 1º de setembro de 2015. Assunto: Orientações sobre os procedimentos para solicitação de alterações na lista de alimentos alergênicos. I. Introdução. A Resolução de Diretoria Colegiada
Art. 1 Esta Resolução possui o objetivo de estabelecer os requisitos mínimos para o registro de medicamentos fitoterápicos.
 RESOLUÇÃO ANVISA Nº 14, DE 31 DE MARÇO DE 2010 Dispõe sobre o registro de medicamentos fitoterápicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere
RESOLUÇÃO ANVISA Nº 14, DE 31 DE MARÇO DE 2010 Dispõe sobre o registro de medicamentos fitoterápicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO RDC Nº 2, DE 17 DE JANEIRO DE 2012
 AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO RDC Nº 2, DE 17 DE JANEIRO DE 2012 Institui o protocolo eletrônico para emissão de Certificado de Registro de Medicamento e Certidão de Registro para
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO RDC Nº 2, DE 17 DE JANEIRO DE 2012 Institui o protocolo eletrônico para emissão de Certificado de Registro de Medicamento e Certidão de Registro para
Os 1 Item(ns) da lista de documentos que não foram cumprido(s):
 da lista de documentos que não foram cumprido(s):") Agência Nacional de Vigilância Sanitária Unidade de Atendimento e Protocolo - UNIAP Listagem de Encaminhamento de Documentação em Caráter Precário Data: 14.11.05 EMPRESA: ANCHIETA INDÚSTRIA E COMÉRCIO
Agência Nacional de Vigilância Sanitária Unidade de Atendimento e Protocolo - UNIAP Listagem de Encaminhamento de Documentação em Caráter Precário Data: 14.11.05 EMPRESA: ANCHIETA INDÚSTRIA E COMÉRCIO
RESOLUÇÃO-RDC N o - 14, DE 31 DE MARÇO DE 2010 DOU Nº 63, 5 de abril de 2010
 RESOLUÇÃO-RDC N o - 14, DE 31 DE MARÇO DE 2010 DOU Nº 63, 5 de abril de 2010 Dispõe sobre o registro de medicamentos fitoterápicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no
RESOLUÇÃO-RDC N o - 14, DE 31 DE MARÇO DE 2010 DOU Nº 63, 5 de abril de 2010 Dispõe sobre o registro de medicamentos fitoterápicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no
RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015
 RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015 Define os requisitos do cadastro de produtos médicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe conferem
RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015 Define os requisitos do cadastro de produtos médicos. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe conferem
RESOLUÇÃO - RDC Nº 23, DE 4 DE ABRIL DE 2012
 RESOLUÇÃO - RDC Nº 23, DE 4 DE ABRIL DE 2012 Dispõe sobre a obrigatoriedade de execução e notificação de ações de campo por detentores de registro de produtos para a saúde no Brasil. A Diretoria Colegiada
RESOLUÇÃO - RDC Nº 23, DE 4 DE ABRIL DE 2012 Dispõe sobre a obrigatoriedade de execução e notificação de ações de campo por detentores de registro de produtos para a saúde no Brasil. A Diretoria Colegiada
Adota a seguinte Consulta Pública e eu, Diretor-Presidente, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 86, de 10 de agosto de 2010. D.O.U de 12/08/2010 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 86, de 10 de agosto de 2010. D.O.U de 12/08/2010 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
adota a seguinte Consulta Pública e eu, Diretor-Presidente, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 71, de 4 de novembro de 2009. D.O.U de 10/11/2009 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 71, de 4 de novembro de 2009. D.O.U de 10/11/2009 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Genéricos - Guia Básico. Autor: Cesar Roberto CRF-RJ: 7461
 Autor: Cesar Roberto CRF-RJ: 7461 Versão 3.00 2001 Introdução: Este guia visa a orientar o profissional farmacêutico sobre os genéricos, e como este deve proceder na hora de aviar uma receita nesta nova
Autor: Cesar Roberto CRF-RJ: 7461 Versão 3.00 2001 Introdução: Este guia visa a orientar o profissional farmacêutico sobre os genéricos, e como este deve proceder na hora de aviar uma receita nesta nova
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA
 1 MINISTÉRIO DA SAÚDE AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO-RDC Nº 2, DE 25 DE JANEIRO DE 2010 (*) Dispõe sobre o gerenciamento de tecnologias em saúde em estabelecimentos de saúde. A Diretoria
1 MINISTÉRIO DA SAÚDE AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA RESOLUÇÃO-RDC Nº 2, DE 25 DE JANEIRO DE 2010 (*) Dispõe sobre o gerenciamento de tecnologias em saúde em estabelecimentos de saúde. A Diretoria
Registro Eletrônico de Medicamentos
 Registro Eletrônico de Medicamentos Emanuela Vieira Gerência Geral de Medicamentos ANVISA - MS Histórico Registro Eletrônico 2008 Contratação da empresa responsável pelo desenho do processo 11/2008 Início
Registro Eletrônico de Medicamentos Emanuela Vieira Gerência Geral de Medicamentos ANVISA - MS Histórico Registro Eletrônico 2008 Contratação da empresa responsável pelo desenho do processo 11/2008 Início
Anvisa - Alimentos - Informes Técnicos
 Página 1 de 7 English Español Institucional Anvisa Publica Serviços Áreas de Atuação Legislação Buscar Informes Técnicos Informe Técnico nº. 36, de 27 de junho de 2008 Orientações sobre a declaração da
Página 1 de 7 English Español Institucional Anvisa Publica Serviços Áreas de Atuação Legislação Buscar Informes Técnicos Informe Técnico nº. 36, de 27 de junho de 2008 Orientações sobre a declaração da
2. Conforme exigido no Anexo II, item 1.4 do edital os produtos devem atender às Normas Regulamentadoras do Ministério do Trabalho e emprego.
 Ilmo. Sr. Dr. Pregoeiro SESI/BA Pregão Eletrônico 20/2012 Objeto: Razões de Recurso IMUNOSUL DISTRIBUIDORA DE VACINAS E PRODUTOS MÉDICOS HOSPITALARES LTDA, já qualificada, em face do Pregão Presencial
Ilmo. Sr. Dr. Pregoeiro SESI/BA Pregão Eletrônico 20/2012 Objeto: Razões de Recurso IMUNOSUL DISTRIBUIDORA DE VACINAS E PRODUTOS MÉDICOS HOSPITALARES LTDA, já qualificada, em face do Pregão Presencial
Diário Oficial da União Seção 1 DOU 11 de dezembro de 2013 [Páginas 76-77]
![Diário Oficial da União Seção 1 DOU 11 de dezembro de 2013 [Páginas 76-77]](/thumbs/21/1240689.jpg "Diário Oficial da União Seção 1 DOU 11 de dezembro de 2013 [Páginas 76-77]") *Este texto não substitui o publicado do Diário Oficial da União* Diário Oficial da União Seção 1 DOU 11 de dezembro de 2013 [Páginas 76-77] RESOLUÇÃO - RDC Nº 54, DE10 DE DEZEMBRO DE 2013 Dispõe sobre
*Este texto não substitui o publicado do Diário Oficial da União* Diário Oficial da União Seção 1 DOU 11 de dezembro de 2013 [Páginas 76-77] RESOLUÇÃO - RDC Nº 54, DE10 DE DEZEMBRO DE 2013 Dispõe sobre
MEDICAMENTOS ESPECÍFICOS
 MEDICAMENTOS ESPECÍFICOS ASPECTOS TÉCNICOS, DÚVIDAS E EXIGÊNCIAS MAIS FREQÜENTES NAS ANÁLISES TÉCNICAS DOS PROCESSOS DE REGISTRO DE MEDICAMENTOS ESPECÍFICOS E FITOTERÁPICOS 1 Objetivos Definir quais são
MEDICAMENTOS ESPECÍFICOS ASPECTOS TÉCNICOS, DÚVIDAS E EXIGÊNCIAS MAIS FREQÜENTES NAS ANÁLISES TÉCNICAS DOS PROCESSOS DE REGISTRO DE MEDICAMENTOS ESPECÍFICOS E FITOTERÁPICOS 1 Objetivos Definir quais são
REGULAMENTO TÉCNICO PARA REGISTRO DE ANTIMICROBIANOS DE USO VETERINÁRIO
 MERCOSUL/GMC/RES. Nº 3/97 REGULAMENTO TÉCNICO PARA REGISTRO DE ANTIMICROBIANOS DE USO VETERINÁRIO TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, as Resoluções Nº 11/93 e 91/93 do Grupo
MERCOSUL/GMC/RES. Nº 3/97 REGULAMENTO TÉCNICO PARA REGISTRO DE ANTIMICROBIANOS DE USO VETERINÁRIO TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, as Resoluções Nº 11/93 e 91/93 do Grupo
RESOLUÇÃO RDC Nº 133, DE 29 DE MAIO DE 2003
 RESOLUÇÃO RDC Nº 133, DE 29 DE MAIO DE 2003 Dispõe sobre o registro de Medicamento Similar e dá outras providências. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição
RESOLUÇÃO RDC Nº 133, DE 29 DE MAIO DE 2003 Dispõe sobre o registro de Medicamento Similar e dá outras providências. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição
adota a seguinte Consulta Pública e eu, Diretor-Presidente Substituto, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 48, de 13 de julho de 2012. D.O.U de 23/07/12 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 48, de 13 de julho de 2012. D.O.U de 23/07/12 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da
RESOLUÇÃO - RDC Nº. 176, DE 21 DE SETEMBRO DE 2006.
 RESOLUÇÃO - RDC Nº. 176, DE 21 DE SETEMBRO DE 2006. Aprova o Regulamento Técnico Contratação de Terceirização para Produtos de Higiene Pessoal, Cosméticos e Perfumes. A Diretoria Colegiada da Agência Nacional
RESOLUÇÃO - RDC Nº. 176, DE 21 DE SETEMBRO DE 2006. Aprova o Regulamento Técnico Contratação de Terceirização para Produtos de Higiene Pessoal, Cosméticos e Perfumes. A Diretoria Colegiada da Agência Nacional
PORTARIA Nº 219/SUMED/ANVISA, DE 23 DE FEVEREIRO DE 2015
 PORTARIA Nº 219/SUMED/ANVISA, DE 23 DE FEVEREIRO DE 2015 A Superintendente de Medicamentos e Produtos Biológicos da Agência Nacional de Vigilância Sanitária no uso das atribuições que lhe confere Portaria
PORTARIA Nº 219/SUMED/ANVISA, DE 23 DE FEVEREIRO DE 2015 A Superintendente de Medicamentos e Produtos Biológicos da Agência Nacional de Vigilância Sanitária no uso das atribuições que lhe confere Portaria
Procedimentos Simplificados para Registro de Medicamentos CLONES
 SUMED Procedimentos Simplificados para Registro de Medicamentos CLONES IV Symposium Sindusfarma IPS/FIP-Anvisa Novas Fronteiras Farmacêuticas nas ciências, tecnologia, regulamentação e sistema de qualidade
SUMED Procedimentos Simplificados para Registro de Medicamentos CLONES IV Symposium Sindusfarma IPS/FIP-Anvisa Novas Fronteiras Farmacêuticas nas ciências, tecnologia, regulamentação e sistema de qualidade
ANEXO. adota a seguinte Resolução de Diretoria Colegiada e eu, Diretor-Presidente, determino a sua publicação:
 Consulta Pública nº 63, de 12 de agosto de 2002. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11, inciso IV, do Regulamento da ANVISA aprovado
Consulta Pública nº 63, de 12 de agosto de 2002. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11, inciso IV, do Regulamento da ANVISA aprovado
RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015. (DOU Seção 1, nº 164, pag. 47, 27.08.2015) (Retificação DOU Seção 1, nº 165, pag. 69, 28.08.
 (Retificação DOU Seção 1, nº 165, pag. 69, 28.08.") RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015 (DOU Seção 1, nº 164, pag. 47, 27.08.2015) (Retificação DOU Seção 1, nº 165, pag. 69, 28.08.2015) Define os requisitos do cadastro de produtos médicos. A
RESOLUÇÃO - RDC Nº 40, DE 26 DE AGOSTO DE 2015 (DOU Seção 1, nº 164, pag. 47, 27.08.2015) (Retificação DOU Seção 1, nº 165, pag. 69, 28.08.2015) Define os requisitos do cadastro de produtos médicos. A
O FOCO DA QUALIDADE NOS PROCESSOS DE TERCEIRIZAÇÃO
 O FOCO DA QUALIDADE NOS PROCESSOS DE TERCEIRIZAÇÃO Grande parte das indústrias farmacêuticas, cosméticos e de veterinários, utilizam processos de terceirização, para otimizar suas produções, para casos
O FOCO DA QUALIDADE NOS PROCESSOS DE TERCEIRIZAÇÃO Grande parte das indústrias farmacêuticas, cosméticos e de veterinários, utilizam processos de terceirização, para otimizar suas produções, para casos
CONSULTA PÚBLICA Nº 01/2012/FUNED
 CONSULTA PÚBLICA Nº 01/2012/FUNED A FUNDAÇÃO EZEQUIEL DIAS, instituída pela Lei 5.594 de 06 de novembro de 1970, regulamentada pelo Decreto nº 45.712, de 30 de agosto de 2011, por intermédio de sua Presidência,
CONSULTA PÚBLICA Nº 01/2012/FUNED A FUNDAÇÃO EZEQUIEL DIAS, instituída pela Lei 5.594 de 06 de novembro de 1970, regulamentada pelo Decreto nº 45.712, de 30 de agosto de 2011, por intermédio de sua Presidência,
WORKSHOP PANORAMA MUNDIAL SOBRE PROBIÓTICOS. Regulamentação atual Medicamentos contendo probióticos
 WORKSHOP PANORAMA MUNDIAL SOBRE PROBIÓTICOS Regulamentação atual Medicamentos contendo probióticos Neemias Silva de Andrade Gerência de Produtos Biológicos / GPBIO Gerência-Geral de Produtos Biológicos,
WORKSHOP PANORAMA MUNDIAL SOBRE PROBIÓTICOS Regulamentação atual Medicamentos contendo probióticos Neemias Silva de Andrade Gerência de Produtos Biológicos / GPBIO Gerência-Geral de Produtos Biológicos,
revoga: Resolução nº 14 de junho de 1978 Resolução nº 15 de abril de 1978 RESOLUÇÃO DE DIRETORIA COLEGIADA - RDC Nº. 268, DE 22 DE SETEMBRO DE 2005.
 título: Resolução RDC nº 268, de 22 de setembro de 2005 ementa não oficial: Aprova o "REGULAMENTO TÉCNICO PARA PRODUTOS PROTÉICOS DE ORIGEM VEGETAL". publicação: D.O.U. - Diário Oficial da União; Poder
título: Resolução RDC nº 268, de 22 de setembro de 2005 ementa não oficial: Aprova o "REGULAMENTO TÉCNICO PARA PRODUTOS PROTÉICOS DE ORIGEM VEGETAL". publicação: D.O.U. - Diário Oficial da União; Poder
CONTRATAÇÃO DE SERVIÇOS DE TERCEIRIZAÇÃO PARA PRODUTOS FARMACÊUTICOS NO ÂMBITO DO MERCOSUL
 MERCOSUL/GMC/RES. Nº 50/02 CONTRATAÇÃO DE SERVIÇOS DE TERCEIRIZAÇÃO PARA PRODUTOS FARMACÊUTICOS NO ÂMBITO DO MERCOSUL TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, as Resoluções Nº
MERCOSUL/GMC/RES. Nº 50/02 CONTRATAÇÃO DE SERVIÇOS DE TERCEIRIZAÇÃO PARA PRODUTOS FARMACÊUTICOS NO ÂMBITO DO MERCOSUL TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, as Resoluções Nº
1.5. Dados pessoais que devem constar na receita médica. 1.6. Validade das receitas de medicamentos antimicrobianos
 Atualizado: 10 / 05 / 2013 FAQ AI 1. Controle de medicamentos antimicrobianos (antibióticos) 1.1. Informações gerais 1.2. Uso contínuo (tratamento prolongado) 1.3. Retenção da segunda via da receita médica
Atualizado: 10 / 05 / 2013 FAQ AI 1. Controle de medicamentos antimicrobianos (antibióticos) 1.1. Informações gerais 1.2. Uso contínuo (tratamento prolongado) 1.3. Retenção da segunda via da receita médica
Perspectivas legais de alegações de propriedades funcionais e ou de saúde
 Perspectivas legais de alegações de propriedades funcionais e ou de saúde 4º Simpósio de Segurança Alimentar Gramado/RS, 29 e 30/05/2012 Antonia Maria de Aquino GPESP/GGALI/ANVISA Missão da Anvisa Promover
Perspectivas legais de alegações de propriedades funcionais e ou de saúde 4º Simpósio de Segurança Alimentar Gramado/RS, 29 e 30/05/2012 Antonia Maria de Aquino GPESP/GGALI/ANVISA Missão da Anvisa Promover
Guia de Submissão Eletrônica de Rótulos
 Agência Nacional de Vigilância Sanitária Guia de Submissão Eletrônica de Rótulos Superintendência de Medicamentos e Produtos Biológicos SUMED www.anvisa.gov.br Brasília, 31 de outubro de 2014. Agência
Agência Nacional de Vigilância Sanitária Guia de Submissão Eletrônica de Rótulos Superintendência de Medicamentos e Produtos Biológicos SUMED www.anvisa.gov.br Brasília, 31 de outubro de 2014. Agência
RESOLUÇÃO CFN N 525/2013
 Página 1 de 7 RESOLUÇÃO CFN N 525/2013 Regulamenta a prática da fitoterapia pelo nutricionista, atribuindo-lhe competência para, nas modalidades que especifica, prescrever plantas medicinais, drogas vegetais
Página 1 de 7 RESOLUÇÃO CFN N 525/2013 Regulamenta a prática da fitoterapia pelo nutricionista, atribuindo-lhe competência para, nas modalidades que especifica, prescrever plantas medicinais, drogas vegetais
RESOLUÇÃO-RDC Nº 17, DE 02 DE MARÇO DE 2007 DOU DE 05/03/2007
 RESOLUÇÃO-RDC Nº 17, DE 02 DE MARÇO DE 2007 DOU DE 05/03/2007 Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento
RESOLUÇÃO-RDC Nº 17, DE 02 DE MARÇO DE 2007 DOU DE 05/03/2007 Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o inciso IV do art. 11 do Regulamento
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 21, DE 28 DE MARÇO DE 2012
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 21, DE 28 DE MARÇO DE
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 21, DE 28 DE MARÇO DE
JAIME CÉSAR DE MOURA OLIVEIRA
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública n 27, de 06 de abril de 2015 D.O.U de 08/04/2015 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública n 27, de 06 de abril de 2015 D.O.U de 08/04/2015 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das
RESOLUÇÃO DE DIRETORIA COLEGIADA - RDC Nº 264, DE 22 DE SETEMBRO DE 2005.
 título: Resolução RDC nº 264, de 22 de setembro de 2005 ementa não oficial: Aprova o "REGULAMENTO TÉCNICO PARA CHOCOLATE E PRODUTOS DE CACAU". publicação: D.O.U. - Diário Oficial da União; Poder Executivo,
título: Resolução RDC nº 264, de 22 de setembro de 2005 ementa não oficial: Aprova o "REGULAMENTO TÉCNICO PARA CHOCOLATE E PRODUTOS DE CACAU". publicação: D.O.U. - Diário Oficial da União; Poder Executivo,
Agência Nacional de Vigilância Sanitária. www.anvisa.gov.br. Consulta Pública n 05, de 28 de janeiro de 2015 D.O.U de 29/01/2015
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública n 05, de 28 de janeiro de 2015 D.O.U de 29/01/2015 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública n 05, de 28 de janeiro de 2015 D.O.U de 29/01/2015 A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 29, DE 12 DE MAIO DE 2008
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 29, DE 12 DE MAIO DE 2008 Aprova o Regulamento
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 29, DE 12 DE MAIO DE 2008 Aprova o Regulamento
Ministério da Agricultura, Pecuária e Abastecimento
 Ministério da Agricultura, Pecuária e Abastecimento D.O.U. Nº 225, sexta-feira, 24 de novembro de 2006. Pág. 10 SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA No- 65, DE 21 DE NOVEMBRO
Ministério da Agricultura, Pecuária e Abastecimento D.O.U. Nº 225, sexta-feira, 24 de novembro de 2006. Pág. 10 SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA No- 65, DE 21 DE NOVEMBRO
Fitoterapia e a prática do Nutricionista. Nutricionista Jacira Santos CRN-2 0091
 Fitoterapia e a prática do Nutricionista Nutricionista Jacira Santos CRN-2 0091 Nutrição Clínica Anos 70 Dietoterapia Hospitalar Anos 80 Dietoterapia em Consultório Anos 90 Fitoquímicos isolados e fitoterápicos
Fitoterapia e a prática do Nutricionista Nutricionista Jacira Santos CRN-2 0091 Nutrição Clínica Anos 70 Dietoterapia Hospitalar Anos 80 Dietoterapia em Consultório Anos 90 Fitoquímicos isolados e fitoterápicos
b) preparado contendo laranja (fruta) e banana (fruta) corresponde a um ingrediente característico;
 preparado contendo laranja (fruta) e banana (fruta) corresponde a um ingrediente característico;") MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO GABINETE DO MINISTRO INSTRUÇÃO NORMATIVA Nº 17, DE 19 DE JUNHO DE 2013 O MINISTRO DE ESTADO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO, no uso da atribuição
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO GABINETE DO MINISTRO INSTRUÇÃO NORMATIVA Nº 17, DE 19 DE JUNHO DE 2013 O MINISTRO DE ESTADO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO, no uso da atribuição
Consulta Pública n. 72, de 14 de julho de 2010. Estabelece os critérios de aceitabilidade de nomes comerciais de medicamentos.
 Consulta Pública n. 72, de 14 de julho de 2010. Estabelece os critérios de aceitabilidade de nomes comerciais de medicamentos. Versão Consolidada n. 07, de 27 de maio de 2013. Para Audiência Pública Resolução
Consulta Pública n. 72, de 14 de julho de 2010. Estabelece os critérios de aceitabilidade de nomes comerciais de medicamentos. Versão Consolidada n. 07, de 27 de maio de 2013. Para Audiência Pública Resolução
Agência Nacional de Vigilância Sanitária ANVISA Gerência-Geral de Tecnologia de Produtos para a Saúde - GGTPS
 Agência Nacional de Vigilância Sanitária ANVISA Gerência-Geral de Tecnologia de Produtos para a Saúde - GGTPS NOTA TÉCNICA N 002/2009/GGTPS/ANVISA 1. Objeto: Orientações para o Peticionamento de Certificado
Agência Nacional de Vigilância Sanitária ANVISA Gerência-Geral de Tecnologia de Produtos para a Saúde - GGTPS NOTA TÉCNICA N 002/2009/GGTPS/ANVISA 1. Objeto: Orientações para o Peticionamento de Certificado
Prescrição e Dispensação de Medicamentos Genéricos e Similares Destaques da Legislação Vigente
 Prescrição e Dispensação de Medicamentos Genéricos e Similares Destaques da Legislação Vigente Brasília, 21 de outubro de 2013. Processo nº: 25351.584974/2013-59 Tema da Agenda Regulatória 2013/2014 nº:
Prescrição e Dispensação de Medicamentos Genéricos e Similares Destaques da Legislação Vigente Brasília, 21 de outubro de 2013. Processo nº: 25351.584974/2013-59 Tema da Agenda Regulatória 2013/2014 nº:
Principais modificações nas normas para registro de medicamentos fitoterápicos
 Principais modificações nas normas para registro de medicamentos fitoterápicos Workshop sobre novas normas de fitoterápicos COFID/GTFAR/GGMED/ANVISA Brasília, 31/05/10 Quem somos PNPMF MS FIOCRUZ MAPA
Principais modificações nas normas para registro de medicamentos fitoterápicos Workshop sobre novas normas de fitoterápicos COFID/GTFAR/GGMED/ANVISA Brasília, 31/05/10 Quem somos PNPMF MS FIOCRUZ MAPA
Serviço Público Federal Conselho Regional de Farmácia do Estado de Santa Catarina - CRF/SC
 Serviço Público Federal Conselho Regional de Farmácia do Estado de Santa Catarina - CRF/SC Trav. Olindina Alves Pereira, 35 - Caixa Postal 472-88020-095 Fone/Fax (48) 222-4702 - Florianópolis - SC. url:
Serviço Público Federal Conselho Regional de Farmácia do Estado de Santa Catarina - CRF/SC Trav. Olindina Alves Pereira, 35 - Caixa Postal 472-88020-095 Fone/Fax (48) 222-4702 - Florianópolis - SC. url:
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 343, DE 13 DE DEZEMBRO DE 2005.
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 343, DE 13 DE DEZEMBRO
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 343, DE 13 DE DEZEMBRO
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA CONJUNTA Nº 32, DE 26 DE OUTUBRO DE 2005
 MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA CONJUNTA Nº 32, DE 26 DE OUTUBRO DE 2005 O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA,
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA CONJUNTA Nº 32, DE 26 DE OUTUBRO DE 2005 O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA,
Políticas de Regulação de Produtos Biotecnológicos
 Diretoria Dirceu Raposo de Melo Gerência Geral de Medicamentos Gerência de Avaliação de Segurança e Eficácia Políticas de Regulação de Produtos Biotecnológicos Daniela Marreco Cerqueira CPBIH/GESEF/GGMED/ANVISA
Diretoria Dirceu Raposo de Melo Gerência Geral de Medicamentos Gerência de Avaliação de Segurança e Eficácia Políticas de Regulação de Produtos Biotecnológicos Daniela Marreco Cerqueira CPBIH/GESEF/GGMED/ANVISA
Unidade de Pesquisa Clínica
 Unidade de Pesquisa Clínica A EQUIVALÊNCIA FARMACÊUTICA NO CONTEXTO DA INTERCAMBIALIDADE ENTRE MEDICAMENTOS GENÉRICOS E DE : BASES TÉCNICAS E CIENTÍFICAS abril/04 SÍLVIA STORPIRTIS1,2; RAQUEL MARCOLONGO1;
Unidade de Pesquisa Clínica A EQUIVALÊNCIA FARMACÊUTICA NO CONTEXTO DA INTERCAMBIALIDADE ENTRE MEDICAMENTOS GENÉRICOS E DE : BASES TÉCNICAS E CIENTÍFICAS abril/04 SÍLVIA STORPIRTIS1,2; RAQUEL MARCOLONGO1;
RESOLUÇÃO RDC ANVISA Nº 345, DE 16 DE DEZEMBRO DE 2002. (D.O.U. de 19/12/02)
") RESOLUÇÃO RDC ANVISA Nº 345, DE 16 DE DEZEMBRO DE 2002 (D.O.U. de 19/12/02) Dispõe sobre a aprovação do Regulamento Técnico para a Autorização de Funcionamento de empresas interessadas em prestar serviços
RESOLUÇÃO RDC ANVISA Nº 345, DE 16 DE DEZEMBRO DE 2002 (D.O.U. de 19/12/02) Dispõe sobre a aprovação do Regulamento Técnico para a Autorização de Funcionamento de empresas interessadas em prestar serviços
Estabelece os requisitos mínimos e o termo de referência para realização de auditorias ambientais.
 RESOLUÇÃO Nº 306, DE 5 DE JULHO DE 2002 Estabelece os requisitos mínimos e o termo de referência para realização de auditorias ambientais. O CONSELHO NACIONAL DO MEIO AMBIENTE-CONAMA, no uso das competências
RESOLUÇÃO Nº 306, DE 5 DE JULHO DE 2002 Estabelece os requisitos mínimos e o termo de referência para realização de auditorias ambientais. O CONSELHO NACIONAL DO MEIO AMBIENTE-CONAMA, no uso das competências
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA Nº 42, DE 31 DE DEZEMBRO DE 2008
 MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA Nº 42, DE 31 DE DEZEMBRO DE 2008 O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA,
MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO SECRETARIA DE DEFESA AGROPECUÁRIA INSTRUÇÃO NORMATIVA Nº 42, DE 31 DE DEZEMBRO DE 2008 O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA,
Amostra grátis de remédios: ANVISA regula a produção e dispensação
 Amostra grátis de remédios: ANVISA regula a produção e dispensação Profª Dra Roseli Calil / DEC Enfº Adilton D. Leite / SADP A ANVISA, através da RDC (RESOLUÇÃO DA DIRETORIA COLEGIADA da Agência Nacional
Amostra grátis de remédios: ANVISA regula a produção e dispensação Profª Dra Roseli Calil / DEC Enfº Adilton D. Leite / SADP A ANVISA, através da RDC (RESOLUÇÃO DA DIRETORIA COLEGIADA da Agência Nacional
Ivo Bucaresky CONBRAFARMA. Diretor ANVISA. Agosto de 2015
 Ivo Bucaresky Diretor ANVISA CONBRAFARMA Agosto de 2015 1 PROGRAMA DE MELHORIA DO PROCESSO DE REGULAMENTAÇÃO Diretrizes: Fortalecimento da capacidade institucional para gestão em regulação Melhoria da
Ivo Bucaresky Diretor ANVISA CONBRAFARMA Agosto de 2015 1 PROGRAMA DE MELHORIA DO PROCESSO DE REGULAMENTAÇÃO Diretrizes: Fortalecimento da capacidade institucional para gestão em regulação Melhoria da
Multivitamínicos Minerais. Regulamentação no Brasil
 Multivitamínicos Minerais Regulamentação no Brasil Workshop sobre Estratégia de Fortificação Caseira no Brasil 29 e 30 de setembro Brasília (DF) Regulamentação Suplementos vitamínicos e ou minerais (Alimentos)
Multivitamínicos Minerais Regulamentação no Brasil Workshop sobre Estratégia de Fortificação Caseira no Brasil 29 e 30 de setembro Brasília (DF) Regulamentação Suplementos vitamínicos e ou minerais (Alimentos)
Agência Nacional de Vigilância Sanitária ANVISA
 Agência Nacional de Vigilância Sanitária ANVISA Coordenação do Comitê Gestor da Implantação do Sistema Nacional de Controle de Medicamentos SNCM Portaria nº 176, de 10/02/2014 NOTA TÉCNICA Nº 01/2015 Considerando
Agência Nacional de Vigilância Sanitária ANVISA Coordenação do Comitê Gestor da Implantação do Sistema Nacional de Controle de Medicamentos SNCM Portaria nº 176, de 10/02/2014 NOTA TÉCNICA Nº 01/2015 Considerando
Deliberação n.º 939/2014, de 20 de março (DR, 2.ª série, n.º 75, de 16 de abril de 2014)
") (DR, 2.ª série, n.º 75, de 16 de abril de 2014) Aprova o formulário de notificação, a efetuar ao INFARMED, I. P., e orientações sobre a prática de reprocessamento de dispositivos médicos de uso único pelo
(DR, 2.ª série, n.º 75, de 16 de abril de 2014) Aprova o formulário de notificação, a efetuar ao INFARMED, I. P., e orientações sobre a prática de reprocessamento de dispositivos médicos de uso único pelo
RESOLUÇÃO ANVISA Nº 22, DE 17 DE JUNHO DE 2010 DOU 18.06.2010
 RESOLUÇÃO ANVISA Nº 22, DE 17 DE JUNHO DE 2010 DOU 18.06.2010 Dispõe sobre a regulamentação da transferência de titularidade de registro de produtos sujeitos à vigilância sanitária em razão de operações
RESOLUÇÃO ANVISA Nº 22, DE 17 DE JUNHO DE 2010 DOU 18.06.2010 Dispõe sobre a regulamentação da transferência de titularidade de registro de produtos sujeitos à vigilância sanitária em razão de operações
http://e-legis.anvisa.gov.br/leisref/public/showact.php?id=10230&word=
 Página 1 de 8 Legislaçã o em Vigilância Sanitária Página Inicial Pesquisa Complementar Estat ísticas do site Normas Consolidadas Publica ções de Hoje Gloss título: Resolução RDC nº 48, de 16 de março de
Página 1 de 8 Legislaçã o em Vigilância Sanitária Página Inicial Pesquisa Complementar Estat ísticas do site Normas Consolidadas Publica ções de Hoje Gloss título: Resolução RDC nº 48, de 16 de março de
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 38, DE 12 DE AGOSTO DE 2013
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 38, DE 12 DE AGOSTO DE 2013 Aprova o regulamento
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO - RDC Nº 38, DE 12 DE AGOSTO DE 2013 Aprova o regulamento
Resolução nº 492 de 26 de novembro de 2008
 Resolução nº 492 de 26 de novembro de 2008 Ementa: Regulamenta o exercício profissional nos serviços de atendimento pré-hospitalar, na farmácia hospitalar e em outros serviços de saúde, de natureza pública
Resolução nº 492 de 26 de novembro de 2008 Ementa: Regulamenta o exercício profissional nos serviços de atendimento pré-hospitalar, na farmácia hospitalar e em outros serviços de saúde, de natureza pública
O Papel da ANVISA na Regulamentação da Inovação Farmacêutica
 O Papel da ANVISA na Regulamentação da Inovação Farmacêutica Renato Alencar Porto Diretor 22 de junho de 2015 Bases legais para o estabelecimento do sistema de regulação Competências na Legislação Federal
O Papel da ANVISA na Regulamentação da Inovação Farmacêutica Renato Alencar Porto Diretor 22 de junho de 2015 Bases legais para o estabelecimento do sistema de regulação Competências na Legislação Federal
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 36, DE 25 DE JULHO DE 2013.
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 36, DE 25 DE JULHO DE
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC Nº 36, DE 25 DE JULHO DE
REGULAMENTO TÉCNICO PARA REGISTRO E FISCALIZAÇÃO DE ESTABELECIMENTOS QUE MANIPULAM PRODUTOS DE USO VETERINÁRIO
 REGULAMENTO TÉCNICO PARA REGISTRO E FISCALIZAÇÃO DE ESTABELECIMENTOS QUE MANIPULAM PRODUTOS DE USO VETERINÁRIO 1. Objetivo Este Regulamento Técnico fixa os requisitos mínimos exigidos para o registro e
REGULAMENTO TÉCNICO PARA REGISTRO E FISCALIZAÇÃO DE ESTABELECIMENTOS QUE MANIPULAM PRODUTOS DE USO VETERINÁRIO 1. Objetivo Este Regulamento Técnico fixa os requisitos mínimos exigidos para o registro e
CONTRATAÇÃO DE TERCEIRIZAÇÃO PARA PRODUTOS DE HIGIENE PESSOAL, COSMÉTICOS E PERFUMES
 MERCOSUL/GMC/RES. Nº 26/06 CONTRATAÇÃO DE TERCEIRIZAÇÃO PARA PRODUTOS DE HIGIENE PESSOAL, COSMÉTICOS E PERFUMES TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, a Decisão Nº 20/02 do Conselho
MERCOSUL/GMC/RES. Nº 26/06 CONTRATAÇÃO DE TERCEIRIZAÇÃO PARA PRODUTOS DE HIGIENE PESSOAL, COSMÉTICOS E PERFUMES TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto, a Decisão Nº 20/02 do Conselho
Frutas e Hortaliças embaladas Aspectos Legais
 Frutas e Hortaliças embaladas Aspectos Legais A embalagem é instrumento de identificação, proteção, movimentação e exposição das frutas e hortaliças frescas. Ela identifica o produto e o seu responsável.
Frutas e Hortaliças embaladas Aspectos Legais A embalagem é instrumento de identificação, proteção, movimentação e exposição das frutas e hortaliças frescas. Ela identifica o produto e o seu responsável.
adota a seguinte Consulta Pública e eu, Diretor Presidente, determino a sua publicação:
 Consulta Pública nº 34, de 28 de junho de 2011. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das atribuições que lhe confere o inciso IV do art. 11 e o art. 35 do Regulamento
Consulta Pública nº 34, de 28 de junho de 2011. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso das atribuições que lhe confere o inciso IV do art. 11 e o art. 35 do Regulamento
Gerência de Produtos Diagnósticos de Uso in vitro
 Gerência de Produtos Diagnósticos de Uso in vitro Brasília, 23 de maio de 2012 Augusto Bencke Geyer Valter Pereira de Oliveira Mônica Cristina A. F. Duarte* Coordenação e Redação Marcella Melo Vergne de
Gerência de Produtos Diagnósticos de Uso in vitro Brasília, 23 de maio de 2012 Augusto Bencke Geyer Valter Pereira de Oliveira Mônica Cristina A. F. Duarte* Coordenação e Redação Marcella Melo Vergne de
ResoluçãodaANVISARDCNº38,de 12 de agosto de 2013: Aprova o regulamento para os programas de. fornecimento de medicamento pósestudo.
 ResoluçãodaANVISARDCNº38,de 12 de agosto de 2013: Aprova o regulamento para os programas de acesso expandido, uso compassivo e fornecimento de medicamento pósestudo. Apresentação: Biól. Andréia Rocha RELEMBRANDO
ResoluçãodaANVISARDCNº38,de 12 de agosto de 2013: Aprova o regulamento para os programas de acesso expandido, uso compassivo e fornecimento de medicamento pósestudo. Apresentação: Biól. Andréia Rocha RELEMBRANDO
PO 001 - GESTÃO DE PROCESSOS E DOCUMENTAÇÃO 008
 1 - OBJETIVO PO 001 - GESTÃO DE PROCESSOS E DOCUMENTAÇÃO 008 Este retrata a forma que deve ser conduzida a gestão dos s da entidade desde a sua concepção até o seu acompanhamento e melhoria. 2 - AUTORIDADE
1 - OBJETIVO PO 001 - GESTÃO DE PROCESSOS E DOCUMENTAÇÃO 008 Este retrata a forma que deve ser conduzida a gestão dos s da entidade desde a sua concepção até o seu acompanhamento e melhoria. 2 - AUTORIDADE
INSTRUÇÃO NORMATIVA CONJUNTA SDA/SDC/ANVISA/IBAMA Nº 1, DE 24 DE MAIO DE 2011.
 INSTRUÇÃO NORMATIVA CONJUNTA SDA/SDC/ANVISA/IBAMA Nº 1, DE 24 DE MAIO DE 2011. O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO - MAPA, o SECRETÁRIO DE DESENVOLVIMENTO
INSTRUÇÃO NORMATIVA CONJUNTA SDA/SDC/ANVISA/IBAMA Nº 1, DE 24 DE MAIO DE 2011. O SECRETÁRIO DE DEFESA AGROPECUÁRIA DO MINISTÉRIO DA AGRICULTURA, PECUÁRIA E ABASTECIMENTO - MAPA, o SECRETÁRIO DE DESENVOLVIMENTO
TENDO EM VISTA: O Tratado de Assunção, o Protocolo de Ouro Preto e as Resoluções Nº 31/97 e 09/01 do Grupo Mercado Comum.
 MERCOSUL/XXXVI SGT Nº11/P. RES. N /11 PROCEDIMENTOS COMUNS PARA AS INSPEÇÕES NOS FABRICANTES DE PRODUTOS MÉDICOS E PRODUTOS PARA DIAGNÓSTICO DE USO IN VITRO NOS ESTADOS PARTES (REVOGAÇÃO DAS RES. GMC Nº
MERCOSUL/XXXVI SGT Nº11/P. RES. N /11 PROCEDIMENTOS COMUNS PARA AS INSPEÇÕES NOS FABRICANTES DE PRODUTOS MÉDICOS E PRODUTOS PARA DIAGNÓSTICO DE USO IN VITRO NOS ESTADOS PARTES (REVOGAÇÃO DAS RES. GMC Nº
adota a seguinte Consulta Pública e eu, Diretor-Presidente, determino a sua publicação:
 Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 56 de 13 de setembro de 2006. D.O.U de 14/09/2006. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
Agência Nacional de Vigilância Sanitária www.anvisa.gov.br Consulta Pública nº 56 de 13 de setembro de 2006. D.O.U de 14/09/2006. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso
MINISTÉRIO DA INDÚSTRIA, DO COMÉRCIO E DO TURISMO
 MINISTÉRIO DA INDÚSTRIA, DO COMÉRCIO E DO TURISMO INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAÇÃO E QUALIDADE INDUSTRIAL - INMETRO Portaria n.º 114, de 29 de junho de 1998. O PRESIDENTE DO INSTITUTO NACIONAL
MINISTÉRIO DA INDÚSTRIA, DO COMÉRCIO E DO TURISMO INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAÇÃO E QUALIDADE INDUSTRIAL - INMETRO Portaria n.º 114, de 29 de junho de 1998. O PRESIDENTE DO INSTITUTO NACIONAL
1. REGISTRO DE ESTABELECIMENTO DE PRODUÇÃO, PREPARAÇÃO, MANIPULAÇÃO, BENEFICIAMENTO, ACONDICIONAMENTO E EXPORTAÇÃO DE BEBIDA E FERMENTADO ACÉTICO.
 ANEXO NORMAS SOBRE REQUISITOS, CRITÉRIOS E PROCEDIMENTOS PARA O REGISTRO DE ESTABELECIMENTO, BEBIDA E FERMENTADO ACÉTICO E EXPEDIÇÃO DOS RESPECTIVOS CERTIFICADOS. 1. REGISTRO DE ESTABELECIMENTO DE PRODUÇÃO,
ANEXO NORMAS SOBRE REQUISITOS, CRITÉRIOS E PROCEDIMENTOS PARA O REGISTRO DE ESTABELECIMENTO, BEBIDA E FERMENTADO ACÉTICO E EXPEDIÇÃO DOS RESPECTIVOS CERTIFICADOS. 1. REGISTRO DE ESTABELECIMENTO DE PRODUÇÃO,
RESOLUÇÃO RDC/ANVISA Nº 161, de 23 de junho de 2004.
 RESOLUÇÃO RDC/ANVISA Nº 161, de 23 de junho de 2004. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11 inciso IV do Regulamento da ANVISA
RESOLUÇÃO RDC/ANVISA Nº 161, de 23 de junho de 2004. A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária, no uso da atribuição que lhe confere o art. 11 inciso IV do Regulamento da ANVISA
RESOLUÇÃO DA DIRETORIA COLEGIADA RDC Nº 45, DE 19 DE SETEMBRO DE 2011. (Alterada pela Resolução RDC n 48, de 25 de setembro de 2014.
 1 RESOLUÇÃO DA DIRETORIA COLEGIADA RDC Nº 45, DE 19 DE SETEMBRO DE 2011. (Alterada pela Resolução RDC n 48, de 25 de setembro de 2014.) D.O.U. de 22/09/2011 Dispõe sobre o regulamento técnico para fórmulas
1 RESOLUÇÃO DA DIRETORIA COLEGIADA RDC Nº 45, DE 19 DE SETEMBRO DE 2011. (Alterada pela Resolução RDC n 48, de 25 de setembro de 2014.) D.O.U. de 22/09/2011 Dispõe sobre o regulamento técnico para fórmulas
REGISTRO DE MEDICAMENTOS
 REGISTRO DE MEDICAMENTOS Deborah Masano Cavaloti Manira Georges Soufia 1 Como a Anvisa avalia o registro de medicamentos novos no Brasil Brasília, 20 de janeiro de 2005 No Brasil, os medicamentos são registrados
REGISTRO DE MEDICAMENTOS Deborah Masano Cavaloti Manira Georges Soufia 1 Como a Anvisa avalia o registro de medicamentos novos no Brasil Brasília, 20 de janeiro de 2005 No Brasil, os medicamentos são registrados
RDC Nº 48, DE 25 DE OUTUBRO DE 2013
 RDC Nº 48, DE 25 DE OUTUBRO DE 2013 ITEM 10 DOCUMENTAÇÕES E REGISTROS Palestrante: Carlos Cezar Martins RDC Nº 48, DE 25 DE OUTUBRO Carlos Cezar Martins DE 2013 Farmacêutico com especialização em Qualidade
RDC Nº 48, DE 25 DE OUTUBRO DE 2013 ITEM 10 DOCUMENTAÇÕES E REGISTROS Palestrante: Carlos Cezar Martins RDC Nº 48, DE 25 DE OUTUBRO Carlos Cezar Martins DE 2013 Farmacêutico com especialização em Qualidade
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA DIRETORIA COLEGIADA RDC N 24, DE 8 DE JUNHO DE 2015
 AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA DIRETORIA COLEGIADA RDC N 24, DE 8 DE JUNHO DE 2015 Dispõe sobre o recolhimento de alimentos e sua comunicação à Anvisa e aos consumidores. A Diretoria Colegiada
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA DIRETORIA COLEGIADA RDC N 24, DE 8 DE JUNHO DE 2015 Dispõe sobre o recolhimento de alimentos e sua comunicação à Anvisa e aos consumidores. A Diretoria Colegiada
PROGRAMA PARA CAPACITAÇÃO DE INSPETORES PARA A VERIFICAÇÃO DO CUMPRIMENTO DAS BOAS PRÁTICAS DE FABRICAÇÃO DE PRODUTOS MÉDICOS
 MERCOSUL/GMC/RES Nº 25/98 PROGRAMA PARA CAPACITAÇÃO DE INSPETORES PARA A VERIFICAÇÃO DO CUMPRIMENTO DAS BOAS PRÁTICAS DE FABRICAÇÃO DE PRODUTOS MÉDICOS TENDO EM VISTA: O Tratado de Assunção, o Protocolo
MERCOSUL/GMC/RES Nº 25/98 PROGRAMA PARA CAPACITAÇÃO DE INSPETORES PARA A VERIFICAÇÃO DO CUMPRIMENTO DAS BOAS PRÁTICAS DE FABRICAÇÃO DE PRODUTOS MÉDICOS TENDO EM VISTA: O Tratado de Assunção, o Protocolo
Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC N 6, DE 30 DE JANEIRO DE 2012
 ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC N 6, DE 30 DE JANEIRO DE
ADVERTÊNCIA Este texto não substitui o publicado no Diário Oficial da União Ministério da Saúde Agência Nacional de Vigilância Sanitária RESOLUÇÃO DA DIRETORIA COLEGIADA - RDC N 6, DE 30 DE JANEIRO DE